GGA+U方法研究ZnO孪晶界对VZn-NO-H复合体对p型导电性的影响∗
2017-08-07吴静静唐鑫龙飞唐壁玉
吴静静唐鑫龙飞唐壁玉
1)(桂林理工大学,有色金属及材料加工新技术教育部重点实验室,桂林 541004)
2)(桂林理工大学材料科学与工程学院,桂林 541004)
3)(广西大学化学化工学院,南宁 530004)
GGA+U方法研究ZnO孪晶界对VZn-NO-H复合体对p型导电性的影响∗
吴静静1)2)唐鑫1)2)†龙飞1)2)唐壁玉3)
1)(桂林理工大学,有色金属及材料加工新技术教育部重点实验室,桂林 541004)
2)(桂林理工大学材料科学与工程学院,桂林 541004)
3)(广西大学化学化工学院,南宁 530004)
(2017年1月24日收到;2017年5月4日收到修改稿)
采用基于密度泛函理论的广义梯度近似平面波赝势方法,探究四种ZnO-Σ7(120)孪晶界中VZn-NO-H复合体的电子结构和p型导电机理.计算结果表明,在ZnO-Σ7(120)孪晶界中,N掺杂后会与锌空位(VZn)、氢填隙(Hi)等点缺陷结合,进而形成VZn-NO-H复合体,并出现在孪晶中的晶格应变集中区.此外,四种孪晶界中孪晶GB7a有利于VZn-NO-H离化能降低,从而使其表现出浅受主特征.分析显示特殊的孪晶结构导致了氮替位(NO)与近邻的O原子间距离缩短,阴离子之间发生相互作用,导致禁带中的空带能级下降,降低了电子跃迁所需能量.这一结果也说明GB7a孪晶界中的VZn-NO-H可能成为N掺杂ZnO材料的p型导电的来源之一.
密度泛函理论,ZnO,孪晶界
1 引 言
氧化锌(ZnO)是一种直接宽带隙半导体材料,其室温下禁带宽度为3.37 eV,激子束缚能为60 meV.ZnO由于其优良的压电和光电特性以及良好的化学稳定性,在透明导电、紫外线发光二极管、太阳能电极材料等诸多领域中具有广泛的应用[1].在ZnO光电性能研究中的关键是p-n结的制备,因此获得稳定的p型ZnO材料是必不可少的.
然而,由于半导体掺杂的不对称性,如何制备高质量且稳定的p型ZnO,一直以来都是ZnO研究的热点之一[2,3].p型ZnO存在自补偿效应、受主低溶解度和高电离能等诸多困难.通常认为NO在p型ZnO是一个潜在的受主,然而理论计算的结果显示了NO是深受主并且离化能高达1.3 eV[4].此外, Hi的自补偿效应对p型掺杂的ZnO影响也较为显著.Gao等[5]在实验中发现N掺杂ZnO在热处理的过程中很容易被Hi所补偿,从而形成No-H复合体.因此,单纯的N替位掺杂ZnO难获得稳定的p型结构.尽管如此,也存在着大量的实验报道证实N掺杂ZnO的确能获得p型导电材料[5-12].因此对于N掺杂获得p型ZnO的问题还存在较大的争议.而对于这个问题的解释,最近Reynolds等[8]在实验中发现,N掺杂的ZnO材料经热处理后,可以获得室温下稳定的p型ZnO,并推论ZnO中的VZn,NO和Hi最终会形成VZn-NO-H复合体且电离能只有130 meV.但Am ini等[13]通过第一性原理计算发现,正常ZnO晶胞内VZn-NO经氢化后形成的VZn-NO-H形成能虽然更低,但同时其受主能级变深,并不是材料p型导电的来源.因此,正常ZnO晶胞内的VZn-NO-H复合体也并不能完全解释实验结果的差异.
ZnO是一种典型的宽禁带半导体,除了点缺陷,还有线缺陷和面缺陷的存在,而这些线缺陷和面缺陷时常易被忽略,但最近的研究显示这些二维或者三维缺陷很大程度上影响着ZnO的物理和化学特性[14-19].而孪晶由于其形成能较低,广泛地存在于异质外延生长中.理论计算结果也显示孪晶对于ZnO中本征点缺陷的形成有着极大的影响,一些孪晶中VZn的形成能仅为-0.19 eV[20],且晶界处的VZn导致界面呈现p型特征[21].从以上研究结果可以发现,相对于正常晶格,孪晶界上原子非规则排列,有利于各种缺陷的形成.而在讨论p型时,若忽略其影响,显然不合理.因而,本文通过建立理论模型来描述孪晶的微观结构,采用密度泛函理论的第一性原理方法计算并讨论ZnO-Σ7(120)孪晶界对VZn-NO-H复合体影响,从热力学原理上分析孪晶界的微观结构和作用机理,从而有效地解释实验中ZnO孪晶界对p型导电性能影响.
2 计算方法
本文采用Vienna ab initio simulation package(VASP)软件中用Perdew-Burke-Ernzerhof泛函描述的密度泛函理论(density functional theory, DFT)框架下的广义梯度近似(generalized gradient approximation,GGA)平面波赝势方法.Zn和O原子的价电子组态分别为:4s23d10和2s2p4.几何优化时,平面波截断能为400 eV;平面波能量的精度为1×10-5eV/atom;迭代过程中作用在每个原子上的力不大于0.01 eV/Å;对Brillouin区的积分计算使用Monkhorst-Pack方案.计算正常ZnO超晶胞时,结构弛豫和电子结构计算的k点选取为3×3×3;计算四种ZnO-Σ7(120)孪晶界时,结构弛豫和电子结构计算的k点分别为1×3×5和3×5×7.计算中使用的正常ZnO超晶胞包含72个原子;四种孪晶界分别为GB7a,GB7b,GB7c和GB7d,其中,GB7a,GB7b和GB7c包含112个原子,GB7d包含108个原子,如图1所示.
由于GGA低估了Zn-3d电子轨道的位置,计算的ZnO带隙值为0.8 eV[22]远小于实验值3.37 eV.为了弥补GGA方法的不足, GGA+U[23-26]常被用于修正Zn-3d电子轨道的位置.虽然GGA+U方法部分修正了计算出的ZnO带隙宽度,但通过计算发现,即使加上一个较高的Ud值,计算出的带隙宽度仍低于实验值,通常在1.4-1.9 eV范围内[21,27-29].这主要是由于ZnO的电子结构不仅与Zn-3d能级有关,而且与O-2p能级有关.当Ud的值增加时,Zn-3d的能级位置逐渐下降,与O-2p能级的p-d排斥作用逐渐减弱,导致价带顶逐渐降低,进而使计算得到的带隙值逐渐增加[28].然而,随着Ud值的增加,3d能带会逐渐地从2p能带中脱离出来,其p-d排斥作用所导致的O-2p能级变化也在减小.在3d能带完全从2p能带中脱离出来之后,若想使计算得出的带隙值达到实验值,对O-2p能带的库仑校正将变得十分必要.因此,最近一些报道提出用GGA+Ud+Up的方法修正ZnO带隙,计算出的带隙值和实验值基本相符[28-32].本文的计算是根据带隙的实验值、Zn和O的电子轨道特征和带隙中缺陷能级的位置,该方法广泛用于GGA+U参数中Ud和Up的拟合[30].结果显示当Ud=12.8 eV和Up=4 eV,且J值为0时,能获得较好的电子结构信息.使用以上参数,计算获得的ZnO带隙值约为3.37 eV,和实验值3.37 eV[16]一致;ZnO的形成焓为-4.00 eV,略大于实验值-3.60 eV[33];Zn-3d轨道的平均能级为-8.80 eV,也在实验值为-8.8--7.5 eV范围内[28];O-2p轨道的展宽为5.19 eV,与实验值-6.0 eV非常接近[7,28].
点缺陷的形成能和离化能在讨论杂质的平衡浓度和体系的热力学稳定方面有着关键的作用.带有q电荷的缺陷α的形成能[34,35]定义为
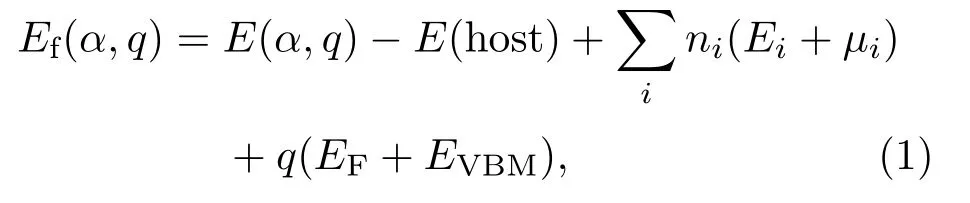
式中E(α,q)为有缺陷且电荷为q时超晶胞的总能量;E(host)则为无缺陷超晶胞的总能量;ni是导致缺陷原子i的数量,如果晶胞中缺陷是由于原子i的增加,则ni<0,若晶胞中缺陷是由于原子i的减少,则ni>0;Ei是固相或气相元素单质的能量,计算出的E(Zn)和E(O)分别是-1.15和-2.79 eV,和实验值-1.30和-2.50 eV相差不大[36];µi是晶胞中所有组分的化学势,µZn和µO遵守平衡条件µZn+µO=ΔHf(ZnO)[35],其中计算出的ZnO形成焓为-4.00 eV.µZn和µO由材料的生长条件控制,锌充足条件时µO=-4.00 eV,µZn=0 eV;氧充足的条件下µO=0 eV,µZn=-4.00 eV.而当缺陷α从q价态转换到q′价态时,其离化能[34]定义为:
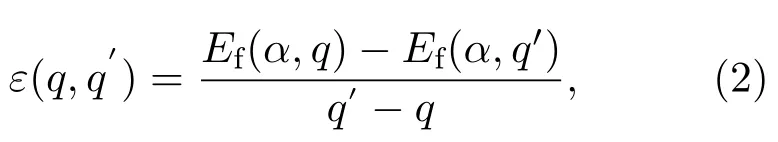
式中Ef(α,q)和Ef(α,q′)分别是带有q和q′电荷的缺陷α的形成能.由于N掺杂ZnO的实验制备过程一般是处于锌充足条件[6-8],所以本文在计算缺陷的形成能和离化能时仅考虑锌充足的情况.
3 计算结果与讨论
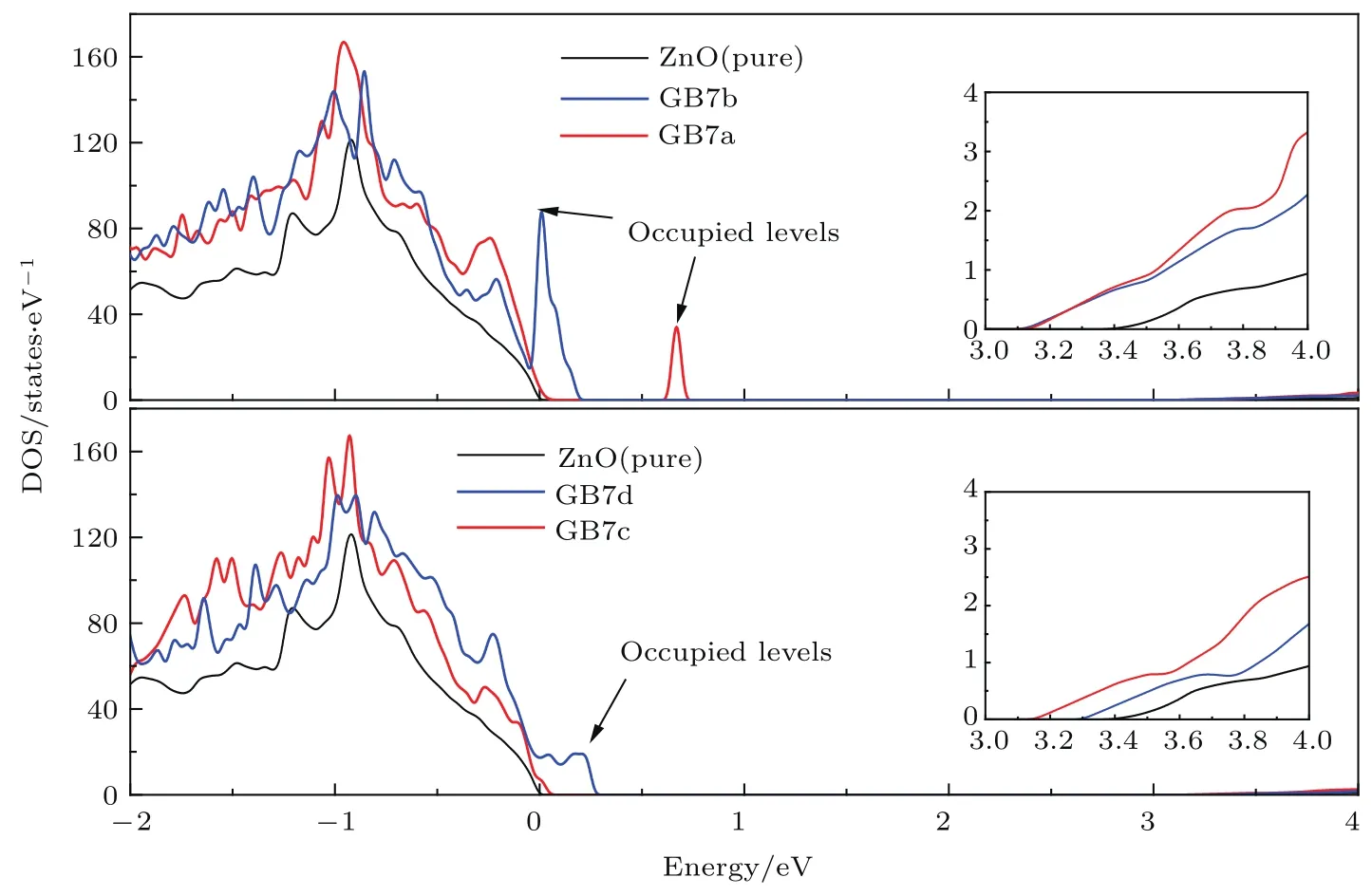
图2 (网刊彩色)纯ZnO,GB7a,GB7b,GB7c和GB7d的DOS,纯ZnO的价带顶设置为能量零点Fig.2.(color on line)DOS of pu re ZnO,GB 7a,GB 7b,GB 7c and GB 7d.The valence band m axim um of bu lk ZnO is chosen to be zero in energy level.
根据文献[37]提出,No通常会和本征VZn结合形成VZn-NO复合体.而ZnO-Σ7孪晶界中VZn-NO复合体可能有两种结合方向:一种为沿c轴方向;另一种为非c轴的方向.经计算后可知,孪晶中以VZn为中心,沿c轴方向VZn-NO复合体的形成能比非c轴方向的平均低0.41 eV.因此,在计算中我们仅考虑VZn-NO复合体沿c轴方向复合的情况.根据对称原则,我们选择了孪晶附近几处典型的位置进行VZn-NO形成能计算,如图1所示,其计算结果在表1中列出.对于含有三配位的孪晶界而言,最低VZn-NO形成能出现在其三配位的位置,即GB7a和GB7b的04和01位置,这主要由于三配位原子仅有三个Zn-O键,当形成VZn时较四配位情况所需的断键更少;对于只含有四配位的孪晶界而言,最低的VZn-NO形成能出现在GB7c的06位置,这主要是由于该位置Zn原子周围的四个Zn-O键相对于晶体内部键长的变化平均为0.04Å,其晶格应变最大,此位置的VZn更有利于应力的释放;对于含有五配位的孪晶界而言,最低VZn-NO形成能并未出现在五配位的01位置,而在其旁边的06位置,弛豫后结果显示,当VZn出现在06位置时,01位置的原子发生偏移并逐渐变为正常四配位结构,也有效地消除了应变.上述结果表明,从能量的角度上看,点缺陷复合体一般常出现在晶界处应变集中区,即原子排列的畸变处.
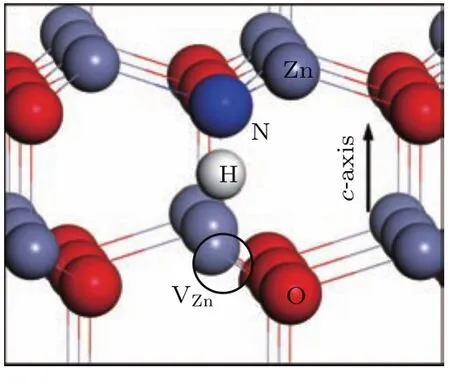
图3 (网刊彩色)正常ZnO晶胞中的VZn-NO-H复合体Fig.3.(color on line)VZn-NO-H com p lex in pu re ZnO supercell.
而近年来的研究显示,N掺杂后ZnO中的VZn-NO复合体会再经氢化最终复合成VZn-NOH[8,13,38],H会吸附在N原子的周围,其结构示意图见图3.因此,根据表1的计算结果,我们分别选择这四种孪晶界中最稳定的VZn-NO进行加H,并计算VZn-NO-H的形成能.表2为计算结果,可以看出,孪晶界中同一位置的VZn-NO-H形成能比VZn-NO的形成能更低.
表1 ZnO-Σ7(120)晶界不同位置处VZn-NO复合体的形成能(锌充足条件)Tab le 1.The form ation energies of VZn-NO com p lex in ZnO-Σ7(120)grain boundary(under Zn-rich cond ition).
表1 ZnO-Σ7(120)晶界不同位置处VZn-NO复合体的形成能(锌充足条件)Tab le 1.The form ation energies of VZn-NO com p lex in ZnO-Σ7(120)grain boundary(under Zn-rich cond ition).
位置 GB 7a/eV GB 7b/eV GB 7c/eV GB 7d/eV 01 3.75 2.41 3.32 3.31 02 3.61 4.15 3.50 3.04 03 3.81 4.03 4.33 4.62 04 1.95 3.25 4.12 2.46 05 3.78 3.01 4.08 3.98 06 3.61 2.77 3.26 2.35 07 - 3.24 3.52 -08 - 3.02 4.33 -
表2中也给出了四种孪晶界中最稳定位置的VZn-NO-H的缺陷离化能.可以看到,除了GB7a,其他三种孪晶界的离化能都有不同程度的提高.GB7b,GB7c和GB7d孪晶界的ε(0/-1),相对于正常ZnO的0.67 eV,分别提升了0.25,0.10和0.27 eV.然而,在GB7a孪晶界中,VZn-NO-H的离化能是降低的,仅为0.38 eV.这主要是由于VZn-NO-H掺杂在GB7a孪晶界的04位上时,复合体中的NO仅与相邻的两个Zn原子成键,并且与07′位上O原子的距离比未掺杂时减少了0.28Å,这导致了两原子的悬挂键之间相互作用增强,从而使复合体中的两个NO-Zn键键能减弱.而在GB7b, GB7c和GB7d孪晶界中,当VZn-NO-H复合体分别掺杂在01,06和06位上时,复合体中的NO和周围O原子的距离平均为2.86,2.85和2.89Å,和正常ZnO晶体内部相差不大,所以其缺陷转变能级较高.GB7a孪晶界的这种特殊结构,导致其中VZn-NO-H的离化能相较其他三种孪晶界中的更低.因此,虽然GB7b,GB7c和GB7d孪晶界和晶体内部的VZn-NO-H呈现深受主特性,但GB7a孪晶界处的VZn-NO-H却主要表现出p型浅受主特征.
图4 (网刊彩色)VZn-NO-H复合体在GB 7a的04位置以及GB 7b的01位置时的DOS,计算获得的费米能级被设置为能量零点Fig.4.(color on line)DOS of VZn-NO-H com p lexes at site 04 in GB 7a and at site 01 in GB 7b,respectively. The Ferm i energy level is chosen to be zero.
图5 (网刊彩色)VZn-NO-H复合体在GB7c的06位置以及GB 7d的06位置时的DOS,计算获得的费米能级被设置为能量零点Fig.5.(color on line)DOS of VZn-NO-H com p lexes at site 06 in GB 7c and GB 7d,respectively.The Ferm i energy level is chosen to be zero.
为了能够更好地比较分析VZn-NO-H在GB7a孪晶界中的浅受主行为,我们分析了复合体在四种孪晶中的DOS,如图4与图5所示.需要指出的是,此时孪晶界处的复合体因VZn的存在而具有铁磁性.由于VZn-NO-H的受主行为主要源于价带顶附近的电子从低能级向高能级的空态跃迁[13],因此,这里主要讨论价带顶附近的态密度特性.从图4(a)可以看到,GB7a孪晶界态密度分布中价带顶附近分别有两个自旋向上的局域峰,主要由N-2p和O-2p电子组成;带隙中有两个自旋向下的局域峰,主要表现为N-2p和O-2p电子特征.而根据另外三种孪晶界态密度分布即图4(b)和图5,可以看出这三种态密度图中都同样有两个上旋局域峰和两个下旋局域峰,并且其能级最低的未占据下旋局域峰分别位于365,424和582 meV,明显高于GB7a孪晶界中的50 meV.这主要是由于复合体掺杂在孪晶界后,N-2p和周围的O-2p的部分电子轨道发生杂化作用,能级发生分裂,进而导致复合体周围的局域自旋电荷分布,而缺陷处的自旋电荷分布很大程度上受到NO和O原子距离影响[20].当复合体位于GB7a孪晶界04位置时,04位置的NO和07位置的O距离拉近,仅为2.38Å,与未掺杂时相比减少了11%,如图6所示.正是由于GB7a孪晶界中这种特殊的结构,导致其NO和O之间的轨道杂化作用较强,如图7(a)所示,其分裂能级正对应态密度图中的自旋局域峰.而复合体掺杂在GB7b和GB7c孪晶界时,晶界处的结构变化不大,其NO和O原子距离仍旧较大.此外,对于GB7d孪晶界而言,虽然未掺杂时06和09位置上的O原子呈现类似O-O键的情况,但掺杂后缺陷处由于应力的释放,会导致01位置O原子的偏移,从而使06和09位置的O原子间距变大,部分电子的相互作用减弱.因此,与GB7a孪晶界相比,上述三种孪晶界中NO和O原子距离较大,导致其N-2p和O-2p的轨道杂化作用较弱,如图7(b)所示.由上述讨论可知,VZn-NO-H掺杂ZnO-Σ7孪晶界后,会导致GB7a孪晶界中缺陷处NO和O原子距离拉近,其轨道杂化作用增强,进而降低了其下旋局域峰的能级,减少了电子跃迁所需的能量;而GB7b,GB7c和GB7d孪晶界中的情形则相反.这正好解释了GB7a孪晶界中VZn-NO-H的浅受主特征,并且与表2中的计算结果相符.
尽管GB7a孪晶界是一个特殊的情况,但基于以上结果,可以认为,在N掺杂ZnO时,由于孪晶界或其他不规则晶界中原子的不规则排列,导致了阴离子直接相互靠近,发生能级杂化成键,改变了原有的禁带中能级的位置,进而有可能降低p型复合体的离化能.因此在讨论N掺杂ZnO或者其他p型掺杂时,不能忽略孪晶及其晶界的影响.
表2 ZnO-Σ7(120)孪晶界中最稳定低位置VZn-NO-H复合体的形成能和离化能(锌充足条件)Tab le 2.The form ation energies and ionization energies of VZn-NO-H com p lex at them ost stab le position in ZnO-Σ7(120)grain boundary(under Zn-rich cond ition).
表2 ZnO-Σ7(120)孪晶界中最稳定低位置VZn-NO-H复合体的形成能和离化能(锌充足条件)Tab le 2.The form ation energies and ionization energies of VZn-NO-H com p lex at them ost stab le position in ZnO-Σ7(120)grain boundary(under Zn-rich cond ition).
位置 Ef/eV ε(0/-1)/eV ε(-1/-2) Bulk ZnO 3.25 0.67 1.64 GB 7a(at 04) 0.52 0.38 1.49 GB 7b(at 01) 0.87 0.92 1.88 GB7c(at 06) 1.62 0.77 1.73 GB 7d(at 06) 0.76 0.94 1.49 /eV

图6 (网刊彩色)GB 7a孪晶界04位置上VZn-NO-H复合体弛豫后的构型Fig.6.(color on line)The con figu ration of VZn-NO-H com p lex at site 04 in GB 7a tw in grain boundary.
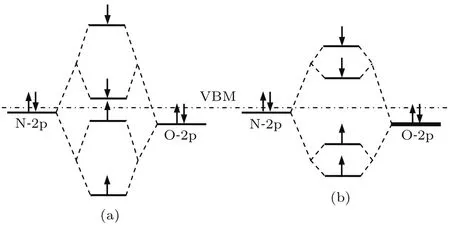
图7 ZnO-Σ7(120)孪晶界处NO-O原子对的能级相互作用图 (a)GB 7a孪晶界;(b)GB 7b,GB 7c和GB 7d孪晶界Fig.7. Schem atic rep resentation of energy level for NO-O pairs in ZnO-Σ7(120)grain boundary: (a)GB 7a grain boundary;(b)GB 7b,GB 7c and GB 7d grain boundary.
4 结 论
采用基于第一性原理计算的GGA+U方法计算了N掺杂ZnO-Σ7(120)孪晶界后形成的VZn-NO及VZn-NO-H复合体.通过讨论其形成能、离化能和态密度分布,发现在ZnO-Σ7孪晶界中这两种复合体更容易出现在孪晶中的晶格应变集中区,即晶界中原子排列的畸变处,且相同位置上VZn-NOH的形成能比VZn-NO更低.虽然VZn-NO-H位于四种ZnO-Σ7孪晶界中都呈现深受主特征,但位于GB7a孪晶界时却有较低的离化能,从而表现出浅受主特征.态密度分布结合GB7a原子结构分析可知,复合体中的NO与邻近的O原子相互作用,其轨道杂化作用增强,进而使禁带中的局域空能级降低,减少了电子跃迁所需的能量,降低了p型VZn-NO-H复合体的离化能.
[1]Özgür U,Alivov Y I,Liu C,Teke A,Reshchikov M A, Doğan S,Av ru tin V,Cho S J,M orkoçH 2005 J.Appl. Phys.98 041301
[2]Fons P,Niki S,Kolobov A V,Ohkubo M,Tom inaga J,Fried rich S,Carboni R,Boscherini F 2006 Nucl.Instrum.M ethods Phys.Res.B 246 75
[3]Chen L J,LiW X,Dai J F,W ang Q 2014 Acta Phys. Sin.63 196101(in Chinese)[陈立晶,李维学,戴剑锋,王青2014物理学报63 196101]
[4]Tarun M C,Iqbal M Z,M cC luskey M D 2011 A IP Adv. 1 022105
[5]Gao J,Zhang X,Sun Y,Zhao Q,Yu D 2010 Nanotechno logy 21 245703
[6]Li X,Yan Y,Gessert T A,Perkins C L,Young D,De-Hart C,Young M,Coutts T J 2003 J.Vac.Sci.Technol. A 21 1342
[7]Lim L Y,Lany S,Chang Y J,Rotenberg E,Zunger A, Toney M F 2012 Phys.Rev.B 86 235113
[8]Reynolds J G,Reynolds C L,M ohanta A,M u th J F, Rowe J E,Everitt H O,Aspnes D E 2013 App l.Phys. Lett.102 152114
[9]Yang T Y,Kong C Y,Ruan H B,Qin G P,Li W J, Liang W W,M eng X D,Zhao Y H,Fang L,Cui Y T 2013 Acta Phys.Sin.62 037703(in Chinese)[杨天勇,孔春阳,阮海波,秦国平,李万俊,梁薇薇,孟祥丹,赵永红,方亮,催玉亭2013物理学报62 037703]
[10]W ang N,Kong C Y,Zhu R J,Q in G P,Dai T L,Nan M,Ruan H B 2007 Acta Phys.Sin.56 5974(in Chinese) [王楠,孔春阳,朱仁江,秦国平,戴特力,南貌,阮海波2007物理学报56 5974]
[11]Tang K,Zhu S,Xu Z,Ye J,Gu S 2017 J.Alloys Com pd. 696 590
[12]Sun JW,Lu Y M,Liu Y C,Shen D Z,Zhang Z Z,Li B H,Zhang J Y,Yao B,Zhao D X,Fan X W 2006 So lid State Comm un.140 345
[13]Am iniM N,Saniz R,Lam oen D,Partoens B 2015 Phys. Chem.Chem.Phys.17 5485
[14]Dom ingos H S,Carlsson JM,B ristowe P D,Hellsing B 2004 Interface Sci.12 227
[15]Erhart P,K lein A,Albe K 2005 Phys.Rev.B 72 085213
[16]Zhang C Y,Li X M,Gao X D,Zhao J L,W an K S,Bian JM 2006 Chem.Phys.Lett.420 448
[17]Lahm er M A,Guergou ri K 2015 M ater.Sci.Sem icond. Process.39 148
[18]Tahir N,K arim A,Persson K A,Hussain S T,Cruz A G,Usm an M,Naeem M,Qiao R,Yang W,Chuang Y D, Hussain Z 2013 J.Phys.Chem.C 117 8968
[19]W ang B,M in J,Zhao Y,Sang W,W ang C 2009 Appl. Phys.Lett.94 192101
[20]Körner W,B ristowe P D,E lsässer C 2011 Phys.Rev.B 84 045305
[21]Li Y H,Xia Q,Guo SK,M a Z Q,Gao Y B,Gong X G, W ei S H 2015 J.App l.Phys.118 045708
[22]Janotti A,van de W alle C G 2007 Phys.Rev.B 76 165202
[23]Sheetz R M,Ponom areva I,Richter E,And riotis A N, M enon M 2009 Phys.Rev.B 80 195314
[24]Hou Q Y,LüZ Y,Zhao C W 2014 Acta Phys.Sin.63 197102(in Chinese)[侯清玉,吕致远,赵春旺2014物理学报63 197102]
[25]Hou Q Y,W u Y,Zhao C W 2014 Acta Phys.Sin.63 137201(in Chinese)[侯清玉,乌云,赵春旺2014物理学报63 137201]
[26]Xu Z C,Hou Q Y 2015 Acta Phys.Sin.64 157101(in Chinese)[许镇潮,侯清玉2015物理学报64 157101]
[27]Bang J,Sun Y Y,West D,M eyer B K,Zhang S 2015 J. M ater.Chem.C 3 339
[28]Agap ito L A,Cu rtarolo S,Buongiorno Nardelli M 2015 Phys.Rev.X 5 011006
[29]W alsh A,Da Silva J L,W ei S H 2008 Phys.Rev.Lett. 100 256401
[30]M a X,W u Y,LüY,Zhu Y 2013 J.Phys.Chem.C 117 26029
[31]Qu L F,Hou Q Y,Xu Z C,Zhao C W 2016 Acta Phys. Sin.65 157201(in Chinese)[曲灵丰,侯清玉,许镇潮,赵春旺2016物理学报65 157201]
[32]Hou Q Y,Dong H Y,M a W,Zhao CW 2013 Acta Phys. Sin.62 157101(in Chinese)[侯清玉,董红英,马文,赵春旺2013物理学报62 157101]
[33]Lim pijum nong S,Li X,Wei SH,Zhang S B 2005 Appl. Phys.Lett.86 211910
[34]Li B S,Feng C,Cui Y X 2010 Chin.Phys.Lett.27 017102
[35]Li H,Schirra L K,Shim J,Cheun H,K ippelen B,M onti O L A,B redas J L 2012 Chem.M ater.24 3044
[36]Lide D R 2014 CRC Handbook ofChem istry and Physics (95th Ed.)(USA:CRC Press)
[37]Liu L,Xu J,W ang D,Jiang M,W ang S,Li B,Zhang Z, Zhao D,Shan C X,Yao B,Shen D Z 2012 Phys.Rev. Lett.108 215501
[38]Yong D Y,He H Y,Tang Z K,W ei S H,Pan B C 2015 Phys.Rev.B 92 235207
(Received 24 January 2017;revised manuscript received 4 May 2017)
Effect of ZnO twin grain boundary on p-type conductivity of VZn-NO-H complex: a GGA + U study∗
Wu Jing-Jing1)2)Tang Xin1)2)†Long Fei1)2)Tang Bi-Yu3)
1)(Key Laboratory ofNew Processing Technology for Nonferrous M etal and Materials,M inistry of Education,Guilin University
of Technology,Guilin 541004,China)
2)(College ofMaterials Science and Engineering,Guilin University of Technology,Guilin 541004,China)
3)(School of Chem istry and Chem ical Engineering,Guangxi University,Nanning 530004,China)
The origin of the p-type conductivity in N-doped ZnOhas been a controversial issuefor years,since isolated N substituted for O site(NO)was found to have high ionization energy.A recent experiment demonstrates that the p-type conductivity is attributed to the VZn-NO-H shallow accep tor com p lex.However,besides the com p lex,there are m any other defects in ZnO,such as tw in grain boundaries.They are comm only two-dim ensional defects,and inevitab ly aff ect the p-type conductivity of the com p lex.By app lying fi rst princip le calculations,we present the electronic structures and p-type conductivity of ZnOΣ7(120)tw in grain boundaries containing VZn-NO-H com p lexes.Four types ofΣ7 tw in grain boundaries are investigated,and the VZn-NO-H com p lex is found to have a tendency to appearing in the stress raisers of the tw in grain boundaries.The lowest formation energy under Zn-rich condition is only 0.52 eV for the com p lex in GB7a,a type ofΣ7 tw in grain boundary with anion-anion bonds,while the value is 3.25 eV for the com p lex in bulk ZnO.For the ionization energy,the com p lex in GB7a ismore easily ionized,and has a value of 0.38 eV, com pared with 0.67 eV in bulk ZnO.The result of density of states show s that the electron transition is dom inated by the em p ty defect levels in forbidden band,which are occupied by O 2p and N 2p orbital.Further analysis indicates that the special structure of GB7a shortens the distances between NOand its neighbor O atom s,and the shortest N—O bond is only 2.38Å,which alsom eans a strong orbital hybridization between O and N.As a result,the energylevelsp litting is enhanced,and the em pty energy level in the forbidden band is shifted down to valence band m aximum.So,GB7a can favor the ionization in VZn-NO-H com p lex.Although GB7a is a special case of the tw in grain boundaries,the result also gives us a new idea to understand the origin of p-type conductivity in N-doped ZnO.
density functional theory,ZnO,tw in grain boundary
PACS:71.15.Mb,77.55.hf,61.72.Mm DO I:10.7498/aps.66.137101
∗国家自然科学基金(批准号:11364009)和广西自然科学基金(批准号:2014GXNSFFA 118004)资助的课题.
†通信作者.E-m ail:x tang@glu t.edu.cn
PACS:71.15.Mb,77.55.hf,61.72.Mm DO I:10.7498/aps.66.137101
*Project supported by the National Natural Science Foundation of China(Grant No.11364009)and GuangxiNatural Science Foundation of China(G rant No.2014GXNSFFA 118004).
†Corresponding author.E-m ail:xtang@glut.edu.cn
