高效液相色谱-四极杆/线性离子阱质谱仪测定盐酸黄酮哌酯中的1-(2-羟乙基)哌啶残留
2017-08-01周长朋郑文凤王春玲
许 洁,周长朋,郑文凤,王春玲
(迪沙药业集团国家认定企业技术中心,山东 威海 264205)
高效液相色谱-四极杆/线性离子阱质谱仪测定盐酸黄酮哌酯中的1-(2-羟乙基)哌啶残留
许 洁*,周长朋,郑文凤,王春玲
(迪沙药业集团国家认定企业技术中心,山东 威海 264205)
建立了盐酸黄酮哌酯中1-(2-羟乙基)哌啶的高效液相色谱-四极杆/线性离子阱质谱(HPLC-QTRAP-MS/MS)定量分析方法。盐酸黄酮哌酯药物用0.1%甲酸溶解后,经Agilent ZORBAX SB-Aq C18(2.1 mm×100 mm,1.8 μm)色谱柱分离,以乙腈-0.1%甲酸水作为流动相进行梯度洗脱,采用电喷雾正离子扫描方式,以选择反应监测(SRM)模式进行检测。结果表明,1-(2-羟乙基)哌啶在10~100 μg/L范围内线性关系良好(r=0.999 7),加标回收率为98.9%~106.8%,相对标准偏差(RSD)均不大于3.0%,方法检出限(S/N=3)为6.0 mg/L,方法定量下限(S/N=10)为20 mg/L。该方法简单、灵敏、准确,适用于盐酸黄酮哌酯中1-(2-羟乙基)哌啶残留的测定。
高效液相色谱-四极杆/线性离子阱质谱仪;盐酸黄酮哌酯;1-(2-羟乙基)哌啶
盐酸黄酮哌酯(Flavoxate hydrochloride,FX),化学名3-甲基黄酮-8-羧酸-2-哌啶乙酯盐酸盐(合成路线见图1),是一种平滑肌松弛药,具有选择性解除泌尿生殖系统平滑肌痉挛的作用,对尿频、尿急、尿痛以及尿失禁症状均有明显的改善作用,临床上常用于治疗下尿路感染、梗阻性疾病以及尿道综合征[1-3]。
液相色谱-质谱联用技术(LC-MS)具有选择性强、灵敏度高、定量准确等优点,近年来已被广泛应用于药品中的杂质研究,其中四极杆/线性离子阱(QTRAP)质谱属于混合型串联质谱,除具有三重四极杆的传统定量功能外,还可进行多级子离子扫描,提供化合物结构信息[10-13]。因此,本文基于高效液相色谱-四极杆/线性离子阱质谱(HPLC-QTRAP-MS/MS)技术,建立了测定盐酸黄酮哌酯药物中1-(2-羟乙基)哌啶的分析方法。该方法操作简单、灵敏度高、回收率好,填补了国内盐酸黄酮哌酯药物中1-(2-羟乙基)哌啶定量检测研究的空白,为盐酸黄酮哌酯药物的质量控制及安全性评价提供了可靠的检测方法。
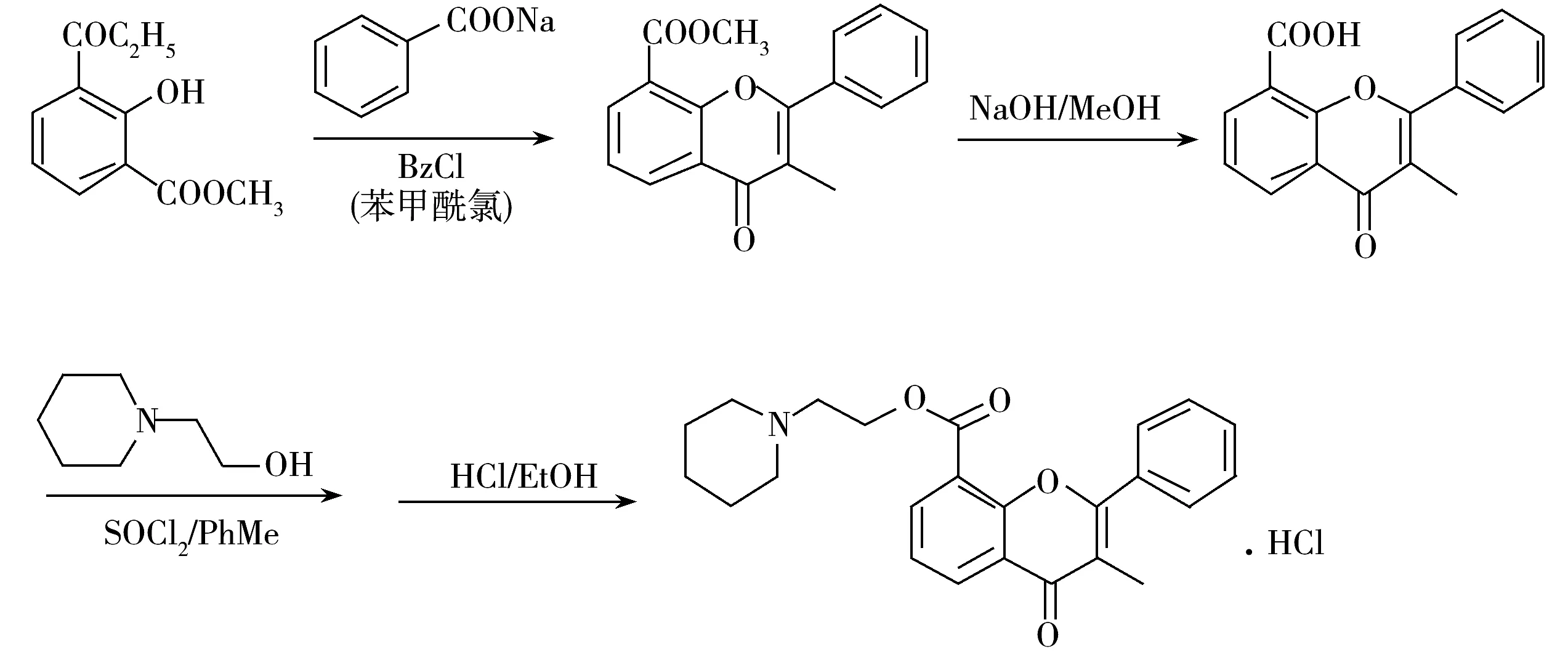
图1 盐酸黄酮哌酯的合成路线图Fig.1 Graphical synthetic route of flavoxate hydrochloride
1 实验部分
1.1 仪器与试剂
Thermo LTQ-XL型高效液相色谱-四极杆/离子阱质谱仪(美国Thermo公司);BT-25S型电子分析天平(瑞士梅特勒-托利多公司);Milli-Q超纯水仪(美国Millipore公司);超声清洗器(昆山超声仪器公司);0.22 μm有机系滤膜(天津津腾实验设备有限公司)。
1-(2-羟乙基)哌啶(纯度≥99%,上海阿拉丁生化科技股份有限公司);甲酸(色谱级,美国Sigma公司);乙腈(色谱级,德国Merck公司);实验用水为Milli-Q超纯水(电阻率为18.25 MΩ·cm);盐酸黄酮哌酯精品(迪沙药业集团有限公司)。
1.2 溶液的配制
对照品溶液:取1-(2-羟乙基)哌啶适量,精密称定,用水溶解并定容,配制成1.0 mg/mL浓度的标准储备液,4 ℃避光保存。取适量标准储备液,用水配制成10.0 mg/L浓度的标准中间液,作为对照品溶液,4 ℃避光保存。上机前用0.1%甲酸水稀释成适当浓度的标准工作溶液,过0.22 μm滤膜,待测。
供试品溶液:取盐酸黄酮哌酯精品适量,精密称定,置于100 mL容量瓶中,加0.1%甲酸水超声溶解并定容,配制成0.1 mg/mL浓度的供试品溶液,过0.22 μm滤膜,待测。
1.3 色谱条件
(2)LVQ神经网络比BP神经网络在进行地下水源热泵系统EER等级划分的预测中速度更快,且稳定性和准确性更高。
色谱柱:Agilent ZORBAX SB-Aq C18(2.1 mm×100 mm,1.8 μm);流动相:0.1%甲酸水溶液(A)和乙腈(B)。采用梯度洗脱模式:0~3.00 min,5%B;3.00~5.00 min,5%~95% B;5.00~8.00 min,95% B;8.01~20.00 min,5% B。流速:0.2 mL/min,柱温:25 ℃;进样体积:10 μL。
1.4 质谱条件
电喷雾离子源(ESI);正离子检测模式;扫描方式:选择反应扫描(SRM)模式;离子源温度:320 ℃;辅助气温度:300 ℃;鞘气:35 psi;辅助气:15 psi;毛细管电压:3 500 V;定量方式:外标法定量;定量离子对:130.0→111.9;碰撞能量(CE):40 V。
2 结果与讨论
2.1 质谱条件的优化
1-(2-羟乙基)哌啶属于极性分子,对其采用ESI离子源比APCI离子源的离子化效率更高,且1-(2-羟乙基)哌啶在水中显碱性,因此采用电喷雾正离子模式(ESI+)进行检测。将1-(2-羟乙基)哌啶配制成1.0 mg/L的标准溶液,先进行一级质谱全扫描,确定其准分子离子[M+H]+峰(m/z130.0),然后对母离子进行子离子全扫描,得到碎片离子,筛选响应值较高、基线噪音低的离子(m/z111.9)作为监测离子,最后对鞘气、辅助气、碰撞能量等参数进行优化,得到“1.4”所述质谱条件。
2.2 配制溶剂的优化
盐酸黄酮哌酯属于酯类化合物,极易发生酯键水解反应,尤其是在碱性溶液中[14-15]。El-Gindy等[16]研究证明,盐酸黄酮哌酯易水解生成3-甲基黄酮-8-羧酸和1-(2-羟乙基)哌啶(图2),在0.1 mol/L NaOH溶液中0.5 h内即可水解完全。本实验考察了盐酸黄酮哌酯在不同配制溶剂中的稳定性,分别用水、水-乙腈(95∶5)、0.1%甲酸水和0.1%甲酸水-乙腈(95∶5) 4种溶液配制得到0.1 mg/mL的盐酸黄酮哌酯样品,分别放置0,0.5,1,2,3 h后上机检测。研究表明,盐酸黄酮哌酯在水和水-乙腈溶液中的水解速度较快,0.5 h内1-(2-羟乙基)哌啶增长近5倍,在0.1%甲酸水和0.1%甲酸水-乙腈溶液中较为稳定,3 h内基本不水解。进一步研究发现,0.1%甲酸水-乙腈配制的样品中1-(2-羟乙基)哌啶的保留能力降低,因此本研究选用0.1%甲酸水作为1-(2-羟乙基)哌啶和盐酸黄酮哌酯供试液的配制溶剂。
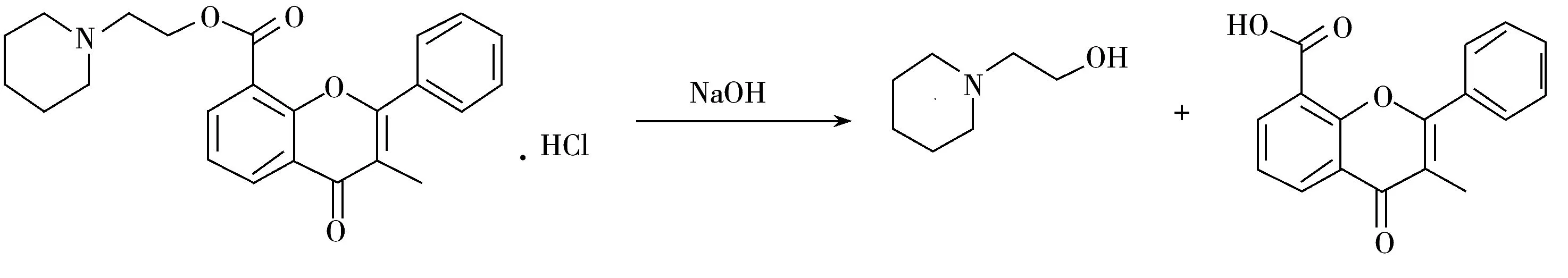
图2 盐酸黄酮哌酯在NaOH溶液中的水解机理Fig.2 Hydrolysis mechanism of flavoxate hydrochloride in sodium hydroxide
2.3 色谱条件的优化
对3种常用的C18色谱柱Agilent Eclipse XDB(2.1 mm×100 mm,3.5 μm)、Thermo Accucore(2.1 mm×100 mm,2.6 μm)和Agilent Zorbax SB-Aq(2.1 mm×100 mm,1.8 μm)进行考察。由于1-(2-羟乙基)哌啶的极性较大,出峰时间较早,在3种色谱柱上均能和盐酸黄酮哌酯主峰达到有效分离,但1-(2-羟乙基)哌啶在Agilent Eclipse XDB柱上的峰形较宽且有拖尾现象,在Thermo Accucore柱上不能有效保留。考虑到本实验的梯度洗脱程序中较长时间为高水相比例,Agilent Zorbax SB-Aq柱具有亲水性表面,可以有效防止固定相的塌陷,在相同检测样品数量下使用寿命明显高于Agilent Eclipse XDB柱和Thermo Accucore柱。另外,Agilent Zorbax SB-Aq柱可使极性化合物在高含水量的流动相中有强保留,从而延长1-(2-羟乙基)哌啶的出峰时间,因此本实验选用Agilent Zorbax SB-Aq柱作进一步的色谱条件优化。
采用正离子扫描模式,在流动相乙腈-水体系中加入适量的甲酸可以提高其离子化效率[17]。本实验考察了流动相A(甲酸水)中分别添加0.02%,0.05%和0.1%甲酸时对1-(2-羟乙基)哌啶峰形、响应强度以及保留时间等方面的影响。结果显示,当流动相A中甲酸含量为0.02%时,虽然1-(2-羟乙基)哌啶的保留时间有所延长,但其峰形较宽而且拖尾严重,响应值较低;提高甲酸含量后,目标化合物的峰形均较好,但在0.1%甲酸条件下其响应值更高,且1-(2-羟乙基)哌啶的保留时间为1.60 min,与盐酸黄酮哌酯主峰(RT=7.1 min)的分离效果较好。因此,本实验最终选择乙腈-0.1%甲酸水溶液作为流动相,采用Agilent Zorbax SB-Aq柱进行分离。
2.4 线性关系、检出限与定量下限
用0.1%甲酸水溶液将10 mg/L的1-(2-羟乙基)哌啶标准溶液稀释后配制成质量浓度分别为10,25,50,75,100 μg/L的系列标准溶液,在最佳检测条件下进行测定,以1-(2-羟乙基)哌啶的峰面积(y)对其质量浓度(x,μg/L)进行线性回归。结果表明,1-(2-羟乙基)哌啶在10~100 μg/L质量浓度范围内线性关系良好(r=0.999 7),线性方程为y=1 396.1+1 161.1x;以3倍信噪比(S/N=3)和S/N=10分别计算方法检出限为6.0 mg/L,定量下限为20 mg/L。
2.5 重复性
按照“1.2”方法平行配制同一批号6份盐酸黄酮哌酯供试品溶液,进行LC-MS测定,考察6份样品中1-(2-羟乙基)哌啶的保留时间和峰面积的相对标准偏差(RSD)。结果显示,6份盐酸黄酮哌酯样品中1-(2-羟乙基)哌啶保留时间的RSD为0.65%,峰面积的RSD为2.6%。方法的重复性良好,可以满足1-(2-羟乙基)哌啶的分析要求。
2.6 样品加标回收率与相对标准偏差
以已知1-(2-羟乙基)哌啶含量的盐酸黄酮哌酯样品的加标回收率考察方法的准确度。在盐酸黄酮哌酯样品中按80%限度、100%限度和120%限度3个水平添加1-(2-羟乙基)哌啶对照品,按照实验方法进行样品前处理和测定,每个加标水平平行测定3个样品。由表1可知,盐酸黄酮哌酯样品中1-(2-羟乙基)哌啶浓度为63.40 mg/L,加标回收率为98.9%~106.8%,RSD均小于3.0%。
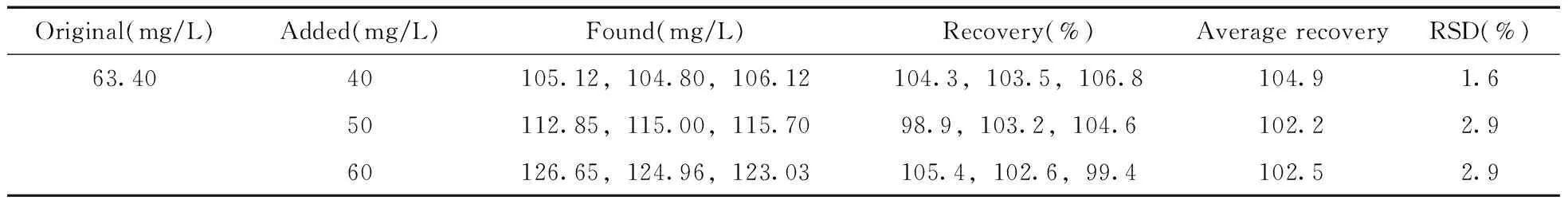
表1 盐酸黄酮哌酯中1-(2-羟乙基)哌啶的加标回收率及相对标准偏差Table 1 Recoveries and RSDs of 1-(2-hydroxyethyl)piperidine ethanol in flavoxate hydrochloride
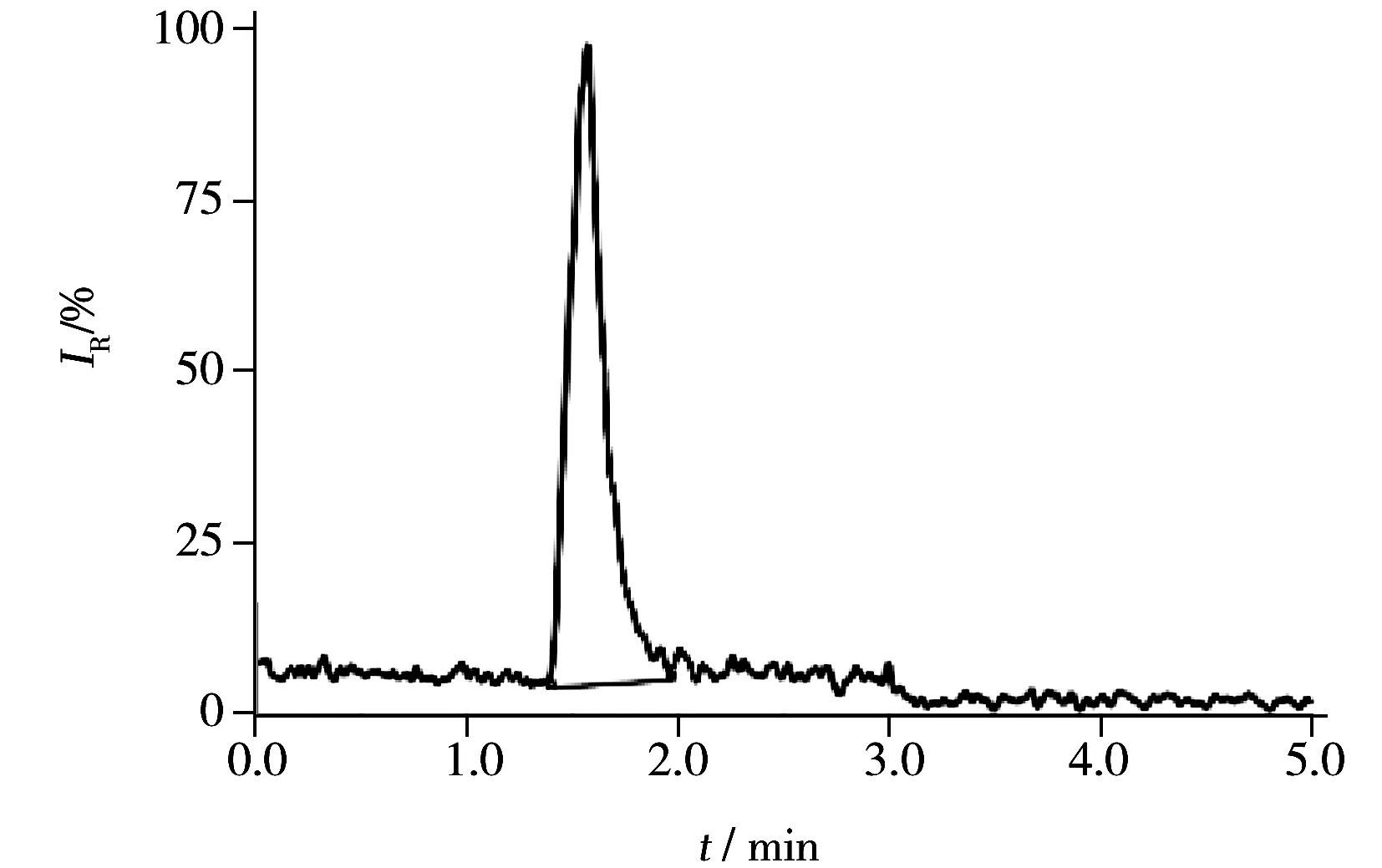
图3 盐酸黄酮哌酯样品中1-(2-羟乙基)哌啶的SRM色谱图Fig.3 SRM chromatogram of 1-(2-hydroxyethyl)piperidine in flavoxate hydrochloride sample
2.7 实际样品的测定
应用本方法对小试3批、中试3批及工艺验证3批共9批盐酸黄酮哌酯样品进行分析测定,所有样品均按照“1.2”方法现用现配。9批样品中均检出1-(2-羟乙基)哌啶残留,含量为53~117 mg/kg,均符合盐酸黄酮哌酯中1-(2-羟乙基)哌啶杂质的限量要求(≤500 mg/kg),图3为某样品的SRM色谱图。
3 结 论
本文首次建立了盐酸黄酮哌酯中1-(2-羟乙基)哌啶残留的HPLC-MS/MS定量检测方法,该方法利用LC-MS/MS的高选择性,有效去除了其他杂质的干扰,确保了方法的高灵敏度,且精密度、回收率和定量下限均能满足限度要求,适用于盐酸黄酮哌酯中1-(2-羟乙基)哌啶残留的定量检测。
[1] Huang Y,Yu Q,Liang M Z,Wang J X,Qin Y P,Zou Y G.J.WestChin.Pharm.Sin.(黄英,余勤,梁茂值,王建新,秦永平,邹远高.华西药学杂志),2000,15(1):9-11.
[2] Zhang X Y,Qiu J X.Chin.J.Clin.Pharmacol.Ther.(张先有, 邱建新.中国临床药理学与治疗学杂志),1996,1(1):37.
[3] Wang Y,Wang T J,Zhang T L.Chin.J.Pharm.Anal.(王玉,王铁杰,张廷兰.药物分析杂志),2002,22(3):202-205.
[4] ICH Guidelines Q3A (R2).ImpuritiesinNewDrugSubstances(新原料药杂质要求),http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3A_R2/Step4/Q3A_R2_Guideline.pdf,2006-10-25.
[5] National Pharmacopoeia Committee.ThePharmacopoeiaofthePeople’sRepublicofChina(Part Ⅳ,2015 Ed.).Beijing:
China Medical Science Press (国家药典委员会.中华人民共和国药典.四部.北京:中国医药科技出版社),2015:9102-9103.
[6] El-Shaheny R N,El-Enany N M,Belal F F.Anal.Methods,2014,6(4):1001-1010.
[7] Zaazaa H E,Mohamed A O,Hawwam M A,Abdelkawy M.Spectrochim.ActaA,2015,134:109-113.
[8] El-Gindy A,Sallam S,Abdel-Salam R A.J.Pharm.Biomed.,2007,44(1):274-278.
[9] Attimarad M.J.Iran.Chem.Soc.,2012,9(4):551-557.
[10] Luo Y Y,Liu X J,Liu X H,Lan C W,Hou Y,Ma Y,Xu L.J.Instrum.Anal.(罗益远,刘秀娟,刘训红,兰才武,侯娅,马阳,徐力.分析测试学报),2015,34(5):519-524.
[11] Zhou C P,Wang D W,Zhang M Y,Xu J,Zheng W F.J.Instrum.Anal.(周长朋,王东武,张曼玉,许洁,郑文凤.分析测试学报),2016,35(9):1111-1115.
[12] Zhang H W,Xu H,Gao J G,Liang C Z,Xu B,Geng J,Wang F M,Zhang X M,Cheng G.Chin.J.Chromatogr.(张鸿伟,许辉,高建国,梁成珠,徐彪,耿娟,王凤美,张晓梅,程刚.色谱),2014,32(6):573-581.
[13] Peng Y W,Yao M C,Luo Z H,Liu G Z.Chin.J.Pharm.Anal.(彭玉薇,姚美村,罗忠华,刘国柱.药物分析杂志),2015,35(5):920-926.
[14] Li W H,Sun C M,Wei Y J.ActaPharm.Sin.(李文红,孙冲梅,魏永巨.药学学报),2015,50(10):1324-1329.[16] El-Gindy A,Abdel-Salam R A.DrugDev.Ind.Pharm.,2008,34 (12):1311-1322.
[17] Li P,Li X L,Miao H,Zhao Y F,Wu Y N.Chin.J.FoodHyg.(李鹏,李晓丽,苗虹,赵云峰,吴永宁.中国食品卫生杂志),2012,24(5):430-435.
Determination of 1-(2-Hydroxyethyl)piperidine in Flavoxate Hydrochloride by HPLC-QTRAP-MS/MS
XU Jie*,ZHOU Chang-peng,ZHENG Wen-feng,WANG Chun-ling
(National Enterprise Technology Center of Disha Pharmaceutical Group,Weihai 264205,China)
A high performance liquid chromatography-quadrupole ion trap tandem mass spectrometric(HPLC-QTRAP-MS/MS) method was developed for the quantification of 1-(2-hydroxyethyl) piperidine residue in flavoxate hydrochloride.After dissolved with 0.1% formic acid,the flavoxate hydrochloride sample was loaded onto an Agilent ZORBAX SB-Aq C18(2.1 mm×100 mm,1.8 μm) column to separate with a mobile phase of 0.1% formic acid-acetonitrile.The electrospray was operated in positive ion mode,and the sample was monitored in the select reaction monitoring(SRM) mode.As a result,the calibration curve of 1-(2-hydroxyethyl)piperidine showed a good linearity(r=0.999 7) in the range of 10-100 μg/L.The recoveries were in the range of 98.9%-106.8%,with relative standard deviations(RSDs) not more than 3.0%.The limit of detection(LOD,S/N=3) for the method was 6.0 mg/L,and the limit of quantitation(LOQ,S/N=10) was 20 mg/L.The method has the advantages of simple operation,high sensitivity and good reproducibility,and could be used for the detection of 1-(2-hydroxyethyl)piperidine in flavoxate hydrochloride.
high performance liquid chromatography-quadrupole ion trap tandem mass spectrometry(HPLC-QTRAP-MS/MS);flavoxate hydrochloride; 1-(2-hydroxyethyl)piperidine
2017-02-22;
2017-03-17
10.3969/j.issn.1004-4957.2017.07.012
O657.63;TQ460.72
A
1004-4957(2017)07-0906-05
*通讯作者:许 洁,硕士,工程师,研究方向:理化检测及药物分析,Tel:0631-3857373,E-mail:xwjxuwenjing1987@126.com
