基于MiSeq分析川中黑山羊瘤胃细菌的多样性及群落结构
2017-07-01陈芸刘旗邓俊良任志华杨颜铱高爽
陈芸,刘旗,邓俊良*,任志华,杨颜铱,高爽
基于MiSeq分析川中黑山羊瘤胃细菌的多样性及群落结构
陈芸1,2,刘旗1,2,邓俊良1,2*,任志华1,3,杨颜铱1,2,高爽1,2
(1.四川农业大学动物医学院,四川成都 611130;2.动物疫病与人类健康四川省重点实验室,四川成都 611130;3.环境公害与动物疾病四川省高校重点实验室,四川成都 611130)
采用MiSeq高通量测序技术分析川中黑山羊瘤胃细菌的多样性及菌群结构。选用3只140日龄健康公羊,其平均体质量为(15.53±0.21) kg,饲喂10 d后,于150日龄时采集瘤胃液(样品A),40 d后再次采集瘤胃液(样品F),提取瘤胃液细菌基因组DNA,对细菌16S rDNA序列V4区进行MiSeq测序。结果显示:1)从样品A与样品F中共获得高质量序列338 830条,聚类后得3 400个运算分类单位(OTU);2)样品A的α多样性指数高于样品F的,但其差异无统计学意义;3)门水平上,样品A最高相对丰度为拟杆菌门的(占总序列数的40.87%),其次为厚壁菌门的(27.19%),样品F最高相对丰度为拟杆菌门的(47.12%),其次为变形菌门的(19.99%),再次为厚壁菌门的(18.05%),样品A厚壁菌门的相对丰度极显著高于样品F的(<0.01);4)在属水平上,样品A与样品F的最高相对丰度均为普雷沃氏菌属的(样品A的为25.54%,样品F的为27.67%),样品A中月形单胞菌属、丁酸弧菌属、瘤胃球菌属、琥珀酸弧菌属、琥珀酸菌属等的相对丰度显著高于样品F的(<0.05)。试验结果表明,川中黑山羊瘤胃中相对丰度最高的菌门为拟杆菌门,相对丰度最高的菌属为普雷沃氏菌属,且两瘤胃样品中部分细菌相对丰度间的差异显著。
川中黑山羊;瘤胃细菌;多样性;群落结构;MiSeq测序;拟杆菌门;普雷沃氏菌属
反刍动物的瘤胃内栖居着大量的微生物,主要包括古细菌、细菌、原虫和真菌,还有少量噬菌体[1],它们在反刍动物的饲料消化和营养代谢中发挥重要作用,如将难消化的植物纤维转换为挥发性脂肪酸、维生素、微生物蛋白等,满足宿主的营养需求[2]。瘤胃内细菌含量可达到微生物总量的95%[3],是瘤胃微生物中最主要的功能类群[4],与宿主的营养状况有直接联系。川中黑山羊是四川中部地区皮肉兼用的地方山羊,主要分布在四川省金堂县、乐至县等,其肉用性能优良,母羊繁殖能力强[5],具有生长快、抗病力强、适应性良好等优点[6–7],是中国现阶段大力发展的优质肉用品种[8]。川中黑山羊1~6月龄时生长发育极快[9],适合于生产肥羔[10]。微生物的多样性和菌群结构与反刍动物的生长有直接联系,但关于川中黑山羊瘤胃微生物菌群结构的研究尚少。笔者采用MiSeq高通量测序技术研究150、190 d(6月龄前后)川中黑山羊的瘤胃微生物多样性和菌群结构,探索瘤胃细菌与川中黑山羊生长性能的关系,旨在为川中黑山羊的临床饲养管理及营养研究提供参考依据。
1 材料与方法
1.1试验动物与饲养管理
选取140日龄、体重约(15.53±0.21)kg的健康雄性(未阉割)川中黑山羊6只,单栏饲喂,混合精料量按体重的2%供给,粗料为新鲜青草,每天08:00和17:00饲喂(饲喂精料后给足青草),全天自由饮水。羊舍温度20 ℃。
1.2样品采集及基因组DNA的提取
饲喂10 d后,随机选取3只,晨饲前以真空泵胃管抽吸法[8]采集瘤胃液(100 mL/只),并将其标记为样品A(150日龄);此后40 d再次采集瘤胃液,并将其标记为样品F (190日龄)。将采集的瘤胃液充分混合均匀后用孔径0.15 mm的 4层灭菌纱布过滤,分装在5 mL 离心管中,迅速置于-70 ℃冰箱中保存。
于每个样品各取2 mL 瘤胃液,经1 000 r/min离心5 min后,取上清液,按细菌基因组DNA提取试剂盒(天根生化科技公司)说明提取DNA,采用微量紫外分光光度计(Nanodrop 2 000)测定DNA浓度及纯度,并采用1%的琼脂糖凝胶电泳检测DNA质量。提取的总基因组DNA于-20 ℃保存,备用。
1.316S rDNA基因的扩增及MiSeq测序
细菌基因组文库构建及上机测序均由上海派森诺科技有限公司完成。以总DNA为模板,对细菌16S rRNA V4区进行PCR扩增,建立DNA文库,所采用的细菌通用引物为520F(5–GCACCTAAYT GGGYDTAAAGNG–3)和802R(5–TACNVGGGTA TCTAATCC–3)。PCR产物进行2%琼脂糖凝胶电泳,用Axygen胶回收纯化试剂盒(AxyPrep DNA Gel Extration Kit,APGX–500)回收V4区扩增产物,并对扩增产物进行定量和质量控制,采用Illumina的TruSeq Nano DNA LT Library Prep Kit构建Illumina测序文库,合格的文库采用MiSeq Reagent Kit V3(600cycles)进行2×300 bp的双端测序。
1.4数据分析
测序原始数据以FASTQ格式保存。首先用FLASH软件(V1.2.7)筛选双端序列,除去错配碱基,获得每个样品的有效序列,再用QIIME软件识别疑问序列(<150 bp或 5端引物错配碱基数>1的序列)。随后,通过QIIME软件(V1.8.0)检查并剔除嵌合体序列,获得每个样品的高质量序列。用QIIME软件对高质量序列按97%的序列相似度进行归并和OTU划分,选取每个OTU中丰度最高的序列作为该OTU的代表序列,并将OUT代表序列与Greengenes数据库(Release 13.8,http://greengenes. secondgenome.com/)的模板序列进行比对,获取每个OTU所对应的分类学信息。用QIIME软件绘制序列数及对应OTU数的稀疏曲线,并分别计算每个样本的α多样性指数(包括Chao指数、ACE指数、Simpson指数和Shannom指数),并获得每个样本在各分类水平的组成。用R软件对属水平的群落组成结构进行PCA分析。采用SPSS Statistics 20统计软件对A样品与F样品菌群的相对丰度进行配对样本检验,以<0.05作为差异显著性判定标准。
2 结果与分析
2.1OTU聚类分析和Alpha多样性分析结果
通过Illumina Miseq高通量测序后,6个瘤胃液样品共获得有效序列373 177条,经质量控制后获得高质量序列338 830条,平均每个样品56 471条,序列平均长度为225 bp。高质量序列按97%相似性归并后,分别获得样品A和样品F的OTU数为1 831、1 569,2个样品共享OTU数为1 311个,共享OUT数分别占样品A、样品F的71.6%、83.55%,表明2组样品中含有大量相同种类的细菌,且样品A中所获得的OTU数高于样品F的。
细菌Alpha多样性用于反映单个样品内部的物种丰富度和均匀度,其中,Chao指数和ACE指数侧重反映样品中物种的丰富度,Shannon指数和Simpson指数侧重反映群落的均匀度。与样品F相比,样品A的丰富度指数(Chao指数和Ace指数)稍高,但其差异无统计学意义(>0.05 );二者多样性指数(Shannon指数和Simpson指数)的差异亦无统计学意义(>0.05)。样品A和样品F的文库覆盖率均在99%以上,表明本试验的测序量可以覆盖各样品大多数微生物,代表了样品的真实情况,能较好地反映瘤胃液中细菌群落种类和结构的多样性。

表1 Alpha多样性指数(n=3)
2.2组间的相似性分析
由基于UniFrac的加权主坐标分析(PCoA)结果(图1)可见,第1主成分的贡献率为33.38%,第2主成分的贡献率为24.90%,第1主成分明显地将样品分成了2部分,样品A分布在图的左边,样品F分布在图的右边,表明样品A和样品F瘤胃细菌群落结构存在一定的差异性。

图1 瘤胃液样品菌群结构Unifrac的加权主坐标分析结果
2.3门水平及属水平上的菌群结构
从样品A和样品F中均鉴定出24个细菌门类。各样品门水平上的菌落组成见图2(相对丰度大于0.1%),两样品的菌群主要来自拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、疣微菌门(Verrucomicrobia)、变形菌门(Proteobacteria)、无壁菌门(Tenericutes)、螺旋体门(Spirochaetes)、蓝细菌门(Cyanobacteria)、TM7、绿弯菌门(Chloroflexi)、纤维杆菌门(Fibrobacteres)、护养菌门(Synergistetes)、黏胶球形菌门(Lentisphaerae)、放线菌门(Actinobacteria)、浮霉菌门(Planctomycetes)和SR1;衣原体门(Chlamydiae)、梭杆菌门(Fusobacteria)、广古菌门(Euryarchaeota)、酸杆菌门(Acidobacteria)等相对丰度低于0.1%的菌门在图2中未列出。样品A中的最优势菌门为拟杆菌门,其相对丰度为40.87%;其次为厚壁菌门,其相对丰度为27.19%。样品F中的最优势菌门也为拟杆菌门,其相对丰度为47.12%;其次为变形菌门(19.99%),再次为厚壁菌门(18.05%),并且样品A中厚壁菌门的相对丰度显著高于样品F的(<0.01)。样品A与样品F相对丰度间存在显著差异的其他菌门见表2。

图2 瘤胃液中相对丰度较高的15个菌门

表2 两样品中相对丰度差异显著或极显著的菌门(n=3)
数据后大写字母示在0.01水平差异显著;数据后小写字母示在0.05水平差异显著。
样品A和样品F中均鉴定出86个细菌菌属。两样品中相对丰度较高的有25个已知菌属(图3),其中相对丰度最高的菌属为普雷沃氏菌属(样品A为25.53%,样品F为27.67%),其次是琥珀酸弧菌属(样品A为7.56%,样品F为3.99%,<0.05),其余菌属的相对含量均较低,如月形单胞菌属 () 、丁酸弧菌属 ()。样品A与样品F间相对丰度差异显著(<0.05)的菌属共有9个,差异极显著(<0.01)的菌属共2个(表3)。样品A和样品F中均发现了大量未分类菌属。这些未分类菌属占总细菌含量的42.15%~47.71%。
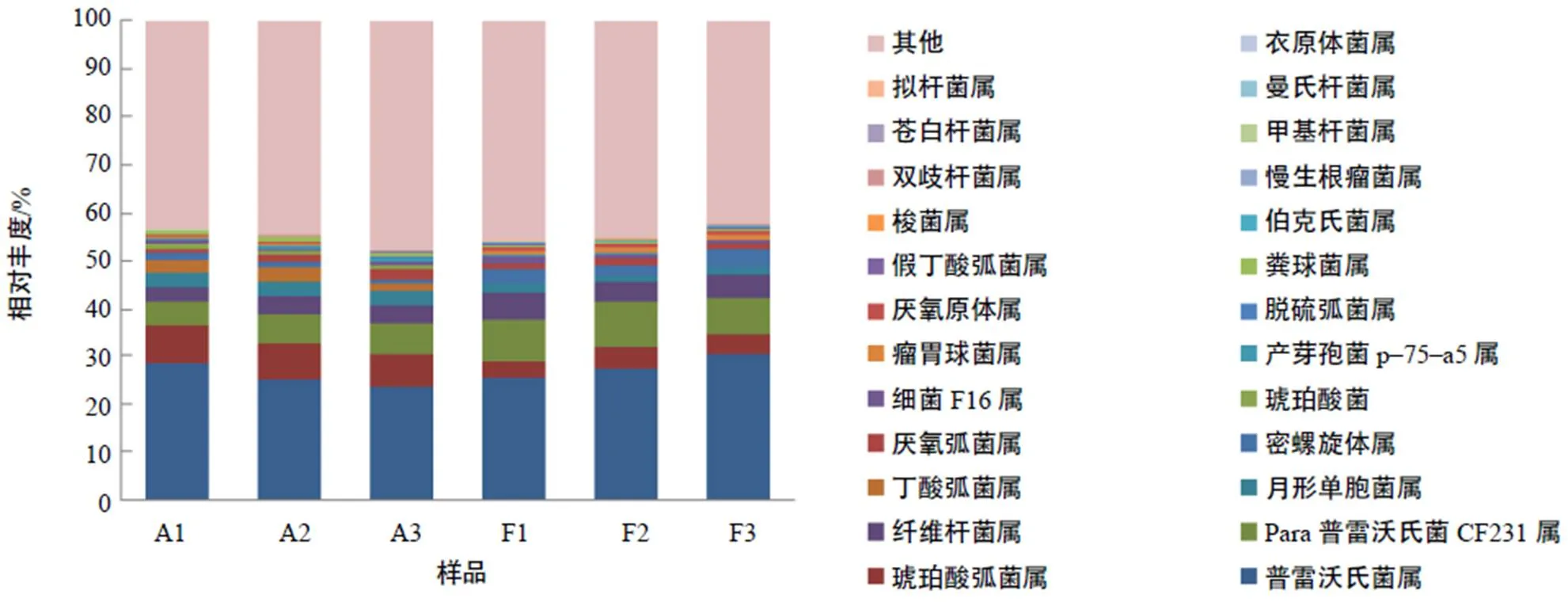
图3 瘤胃液中相对丰度较高的25个菌属

表3 两样品中相对丰度差异显著或极显著的菌属(n=3)
3 讨论与结论
反刍动物瘤胃微生物群落对宿主的健康发挥重要作用,其结构与组成常被用于评估宿主的健康状况[11]。本研究中150 日龄川中黑山羊瘤胃中细菌的多样性高于190日龄的,但二者的差异无统计学意义,其原因可能是组内方差过大。
本研究中,拟杆菌门、厚壁菌门和变形菌门为川中黑山羊瘤胃中的优势菌门。该结果与反刍动物瘤胃内优势菌门研究结果[12–14]一致,这3种菌门在植物纤维降解过程中起重要作用[15]。本试验中川中黑山羊瘤胃细菌相对丰度最高的为拟杆菌门(样品A为40.87%,样品F为47.12%),但绵羊瘤胃内相对丰度最高的为厚壁菌门(44.37%)[16],蒙古羊瘤胃相对丰度最高的为拟杆菌门(67%)[17],表明不同品种羊瘤胃内的优势细菌相对丰度存在一定的差异。
川中黑山羊瘤胃中细菌的最优势菌属为普雷沃氏属,这与针对其他品种山羊及其他反刍动物进行研究的结果[18]一致,这可能与普雷沃氏菌属具有多重功能及多种基因型[19]有关。普雷沃氏菌属中布氏普雷沃氏菌、普雷沃氏短杆菌和栖瘤胃普雷沃氏菌等均具有蛋白酶活性和纤维素酶基因[20],日粮中添加淀粉会使普雷沃氏菌丰度升高[21](主要由于普雷沃氏菌属产生大量复合酶[22]加速了淀粉的降解)。
本研究中150 日龄川中黑山羊瘤胃中厚壁菌门细菌的含量极显著高于190 日龄的(<0.01),这与文献[23]的研究结果相似。韩旭峰[23]发现,拟杆菌门细菌的相对丰度与日龄呈正相关,而厚壁菌门和互养菌门细菌的相对丰度随日龄的增长呈下降趋势。本研究中,150 日龄川中黑山羊瘤胃中厚壁菌门月形单胞菌属、丁酸弧菌属、琥珀酸菌属的相对丰度显著高于190 日龄的。该结果与文献[23]的结果一致。150 日龄川中黑山羊瘤胃中变形菌门琥珀酸弧菌属细菌的相对丰度极显著高于190日龄的(<0.01),说明二者的主要差异来自于厚壁菌门和变形菌门。胃肠道内厚壁菌门的相对含量与机体的肥胖成正相关。该菌门相对丰度的增加与机体对能量的摄入呈正相关[24],且厚壁菌门中丁酸弧菌属、琥珀酸菌属细菌为瘤胃内重要的纤维降解菌,变形菌门中琥珀酸弧菌属细菌为半纤维素降解菌[25]。由此可推测,这几种功能菌相对丰度的降低可能会降低瘤胃对植物纤维的分解代谢,从而影响瘤胃的消化功能及宿主生长性能[26]。1~6月龄川中黑山羊的平均生长速率大于6月龄以上黑山羊的[10],可见,厚壁菌门相对丰度降低是6月龄以上川中黑山羊生长减缓的主要原因。
综上所述,川中黑山羊瘤胃细菌多样性随日龄增长有所降低,但150日龄的与190日龄的差异无统计学意义;瘤胃细菌相对丰度最高的菌门为拟杆菌门,相对丰度最高的菌属为普雷沃氏菌属;厚壁菌门相对丰度的降低与川中黑山羊生长速率有一定的相关性。
[1] E EDWARDS J,R MCEWAN N,J TRAVIS A,et al. 16S rDNA library–based analysis of ruminal bacterial diversity[J].Antonie Van Leeuwenhoek,2004,86(3):263–281.DOI:10.1023/B:ANTO.0000047942.69033.24.
[2] JOSE V L,APPOOTHY T,MORE R P,et al. Metagenomic insights into the rumen microbial fibrolytic enzymes in Indian crossbred cattle fed finger millet straw[J].AMB Express,2017,7(1):13.DOI:10.1186/ s13568–016–0310–0.
[3] MACKIE R I,AMINOV R I,WHITE B A,et al. Molecular ecology and diversity in gut microbial ecosystems[J].Ruminant Physiology:Digestion,Metabolism,Growth and Reproduction,2000,2:61–77.DOI:10.1079/9780851994635.0061.
[4] SHI P J,MENG K,ZHOU Z G,et al.The host species affects the microbial community in the goat rumen[J]. Lett Appl Microbiol,2008,46(1):132–135.DOI:10.1111/j.1472–765X.2007.02274.x.
[5] 文永照.川中黑山羊(乐至型)繁殖性能研究[C]//第九届羊业发展大会论文集,2012:88–92.
[6] 龙石太,胡亮,吴宪红,等.川中黑山羊、基因 cDNA 克隆及生物信息学分析[J].江苏农业科学,2014,42(3):19–22,23.
[7] 杨珂伟,字向东,付锡三,等.乐至黑山羊的同期发情研究[J].西南民族大学学报(自然科学版),2016,42(2):151–155.DOI:10.11920/xnmdzk.2016.02.005.
[8] 周多恩,刘德武,廖迎新,等.湖羊和川中黑山羊在华南地区的适应性研究[J].华南农业大学学报,2016,37(5):19–23.
[9] 陈建,何焕周,姜栾平,等.乐至黑山羊优良性状的研究[J].中国草食动物,2007(增刊1):45–46.DOI:10.3969/j.issn.2095–3887.2007.z1.015.
[10] 文永照.乐至黑山羊品种研究报告[J].中国草食动物,2004(增刊1):116–119.DOI:10.3969/j.issn.2095–3887. 2004.z1.056.
[11] KOCHERGINSKAYA S A,AMINOV R I,WHITE B A.Analysis of the rumen bacterial diversity under two different diet conditions using denaturing gradient gel electrophoresis,random sequencing,and statistical ecology approaches[J].Anaerobe,2001,7(3):119–134.DOI:10.1006/anae.2001.0378.
[12] KIM M,MORRISON M,YU Z.Status of the phylogenetic diversity census of ruminal microbiomes[J]. FEMS Microbiol Ecol,2011,76(1):49–63.DOI:10.1111/j.1574–6941.2010.01029.x.
[13] JAMI E,MIZRAHI I.Composition and similarity of bovine rumen microbiota across individual animals[J]. PLoS ONE,2012,7(3):e33306.DOI:10.1371/journal. pone.0033306.
[14] PETRI R M,SCHWAIGER T,PENNER G B,et al. Characterization of the Core Rumen Microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge[J].PLoS ONE,2013,8(12):e83424.DOI:10.1371/journal.pone. 0083424.
[15] 李亚丹,任宏伟,吴彦彬,等.拟杆菌与肠道微生态[J].微生物学通报,2008,35(2):281–285.DOI:10.3969/j.issn.0253–2654.2008.02.023.
[16] 曾燕.成年健康绵羊胃肠道菌群的研究[D].成都:四川农业大学,2015.
[17] 曾燕,简平,倪学勤,等.Illumina MiSeq测序平台测定蒙古羊瘤胃液相和固相菌群多样性[J].动物营养学报,2015,27(10):3256–3262.DOI:10.3969/j.issn.1006– 267x.2015.10.034.
[18] CUNHA I S,BARRETO C C,COSTA O Y,et al. Bacteria and Archaea community structure in the rumen microbiome of goats () from the semiarid region of Brazil[J].Anaerobe,2011,17(3):118–124.DOI:10.1016/j.anaerobe.2011.04.018.
[19] AVGUSTIN G,WALLACE R J,FLINT H J.Phenotypic diversity among ruminal isolates of:proposal ofsp.nov.,sp.nov.,andsp.nov.and redefinition of[J].Int J Syst Bacteriol,1997,47(2):284–288.DOI:10.1099/00207713–47–2–284.
[20] PURUSHE J,FOUTS D E,MORRISON M,et al. Comparative genome analysis ofand:insights into their environmental niche[J].Microbial Ecology,2010(4):721–729.DOI:10.1007/s00248–010–9692–8.
[21] ZENED A,COMBES S,CAUQUIL L,et al.Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets[J].FEMS Microbiol Ecol,2013,83(2):504–514.DOI:10.1111/1574–6941.12011.
[22] GARDNER R G,WELLS J E,RUSSELL J B,et al.The cellular location ofbeta–1,4–D–endoglucanase and its occurrence in other strains of ruminal bacteria[J].Appl Environ Microbiol,1995,61(9):3288–3292.
[23] 韩旭峰.日龄、日粮精粗比对陕北白绒山羊瘤胃微生物区系影响的研究[D].杨凌:西北农林科技大学,2015.
[24] LEY R E,TURNBAUGH P J,KLEIN S,et al.Microbial ecology:human gut microbes associated with obesity[J]. Nature,2006,444(7122):1022–1023.DOI:10.1038/ 4441022a.
[25] 王继文,王立志,闫天海,等.山羊瘤胃与粪便微生物多样性[J].动物营养学报,2015,27(8):2559–2571. DOI:10.3969/j.issn.1006–267x.2015.08.030.
[26] PETRI R M,FORSTER R J,YANG W,et al. Characterization of rumen bacterial diversity and fermentation parameters in concentrate fed cattle with and without forage[J].J Appl Microbiol,2012,112(6):1152–1162.DOI:10.1111/j.1365–2672.2012.05295.x.
责任编辑:王赛群
英文编辑:王库
Analysis the bacterial diversity in rumen and their community structure from Chuanzhong black goat using MiSeq sequencing technology
CHEN Yun1,2,LIU Qi1,2,DENG Junliang1,2*,REN Zhihua1,3,YANG Yanyi1,2,GAO Shuang1,2
(1.College of Veterinary Medicine, Sichuan Agriculture University, Chengdu 611130,China; 2.Key Laboratory of Animal Disease and Human Health of Sichuan Province, Chengdu 611130,China; 3.Key Laboratory of Environmental Hazards and Animal Diseases of Sichuan Province Colleges and Universities, Chengdu 611130,China)
The study was designed to reveal the bacterial diversity in rumen and their community structure from Chuanzhong black goatusing MiSeq sequencing technology. Three140–day–old and weight of (15.53±0.21)kg Chuanzhong black goats were selected for collecting rumen liquid after normal feeding of 10 d (sample A),after 40 d ,the rumen fluid was collected again(sample F), respectively. Total DNA were extracted for amplifying V4 area in 16S rDNA, and their products were sequenced by Illumina MiSeq sequencing system. The results showed that: 1) A total of 338 830 high quality valid sequences from the sample A and the sample F, 3 400 operational taxonomic units were obtained. 2) The alpha diversity index of sample A was higher than that of sample F, however, their difference did not reach to statistical significance level. 3) At phylum level, the most abundant phylum in sample A was Bacteroidetes (accounted for 40.87% of the total sequences), followed by Firmicutes (it accounted for 27.19% of the total); the most abundant phylum in sample F was Bacteroidetes (accounted for 47.12% of the total sequences) followed by Proteobacteria(it accounted for 19.99% of the total), and Firmicutes(it accounted for 18.05% of the total); the relative abundance of Firmicutes in sample A was very significantly higher than that in sample F (<0.01). 4) At genus level, the most abundant genera was(sample A was 25.54%, sample F was 27.67%); the relative abundance of,,,andin sample A was significantly higher than that in sample F (<0.05). In conclusion,was the most abundant phylum in the rumen bacterial community of Chuanzhong black goats, andwas the most abundant genus; there were significant difference between sample A and sample F.
Chuanzhong black goat; rumen bacteria; diversity; community structure; MiSeq sequencing;Bacteroidetes;
10.13331/j.cnki.jhau.2017.03.012
S826.8
A
1007-1032(2017)03-0286-06
2017–01–26
2017–04–08
“长江学者和创新团队发展计划”创新团队项目( IRT0848);四川农业大学“双支”计划项目(03572070)
陈芸(1992—),女,云南昭通人,硕士研究生,主要从事反刍动物消化道微生物研究,609835279@qq.com;
,邓俊良,博士,教授,主要从事营养代谢病及中毒病研究,dengjl213@126. com
投稿网址:http://xb.hunau.edu.cn
