一对双方染色体异常夫妇的遗传学分析与产前诊断
2017-06-19杨育青
杨育青

[摘要] 目的 对一对有不良孕产史且双方染色体异常的夫妇进行染色体遗传学分析和产前诊断并提供遗传咨询。方法 2016年1月抽取夫妻双方的外周血及2016年4月孕妇的羊水标本进行常规细胞培养和染色体制备,并综合应用染色体核型分析(G显带)、荧光原位杂交;单核苷酸多态性微阵列进行染色体遗传学分析。 结果 孕妇染色体核型:46,XX,del(X) (p22.2-pter),为Xp部分缺失携带者,其染色体在Xp22.33p22.2区段存在16.57 Mb片段的缺失;丈夫染色体核型:46,XY,inv(9)(p11q13)为染色体9号臂间易位携带者;胎儿染色体核型为:46,XX。 结论 夫妻可生育正常后代,如再次怀孕仍需行产前诊断。
[关键词] 染色体核型分析;产前诊断;荧光原位杂交;单核苷酸多态性微阵列
[中图分类号] R714.55 [文献标识码] A [文章编号] 1674-0742(2017)04(c)-0041-03
[Abstract] Objective To conduct the genetic analysis and prenatal diagnosis of a couple with chromosomal abnormalities and provide genetic consultation. Methods The peripheral blood of the couple extracted in January 2016 and amniotic fluid specimen of delivery women in April 2016 were selected for the routine cell culture and chromosome preparation, and the chromosomal genetic analysis was conducted by the chromosomal karyotypes (G banding), fluorescence in situ hybridization and SNP array. Results Chromosomal karyotypes of delivery women, 46,XX,del(X) (p22.2-pter), Xp partial deletion carrier, and the chromosome had the 16.57 Mb segment deletion in the Xp22.33p22.2 section; chromosomal karyotypes of husband, 46,XY,inv(9)(p11q13), No.9 pericentric translocation carrier, and the chromosomal karyotypes of fetus; 46,XX. Conclusion The couple can produce the normal offspring, and it is necessary to receive the prenatal diagnosis if the wife was pregnant again.
[Key words] Chromosomal karyotypes analysis; Prenatal diagnosis; Fluorescence in situ hybridization; Single nucleotide polymorphisms
染色體病在遗传病中占有相当的比重,发病率大约为5‰。其中由X性染色体异常引起的疾病发病率很高,其临床表现有性腺发育不全、智力低下、两性畸形、原发或继发不孕等。而常染色体9号臂间倒位则是最常见的染色体结构变异之一,目前大部分学者认为9号倒位会引起自然流产、死胎、不孕不育等。该研究对象为2016年来该院诊治的一对夫妇,孕妇为Xp部分缺失携带者,丈夫为染色体9号臂间倒位携带者,这种情况比较少见,现将对二者的遗传学分析与产前诊断报道如下。
1 对象与方法
1.1 研究对象
张某30岁,孕8周,因两年前有一次自然流产,与丈夫前来该院遗传咨询中心就诊。孕妇查体:身高155 cm,四肢骨骼发育正常,性发育正常,17岁初潮,月经周期30 d,月经规律, 智力正常。丈夫查体正常。经咨询并知情同意后夫妇先行外周血染色体检查。孕妇待到合适孕周后加行产前诊断及非孕期性激素、甲状腺功能检测。
1.2 方法
1.2.1 孕妇张某 ①外周血淋巴细胞培养、收获和制片:抽取静脉血3 mL,取1 mL接种于外周血培养基进行培养,72 h后按实验室操作规范进行收获和制片,将部分细胞悬液置-20℃保存,将染色体片常规G显带;取2 mL静脉血于EDTA抗凝管,提取基因组DNA。②羊水细胞培养:于孕18周经B 超引导行羊膜腔腹穿刺抽取羊水2 mL,换注射器针头再取羊水20 mL分两线进行接种培养;羊水细胞培养一周后换液,视克隆生长情况按实验室操作规范进行收获和制片,将染色体片常规G显带;染色体核型分析:镜下计数20个细胞并分析5个,遇到嵌合时按全国行业标准进行处理,最后根据ISCN2005报告染色体核型。③SNP-array( AffymetrixCytoScan 750K Array)测定:按仪器试剂厂家提供的实验手册进行实验,用分析平台Affymertrix GeneChip 750K SNP 进行实验数据分析。④FISH 验证:将步骤①制备的细胞悬液,通过滴片、热变性处理,按照FISH实验方法与选定的XYpter(绿色)/XYqter(红色)双色探针进行杂交,经洗片复染后进行结果分析。⑤在非孕期时抽取静脉血3 mL,用UniCel DxI 800全自动化学发光分析仪及其相应试剂检测性激素六项及甲状腺功能,实验操作按试剂说明书进行。
1.2.2 丈夫 ①外周血淋巴细胞培养、收获、制片及核型分析同上;②C带分析按实验室操作规范进行。
2 结果
2.1 孕妇张某
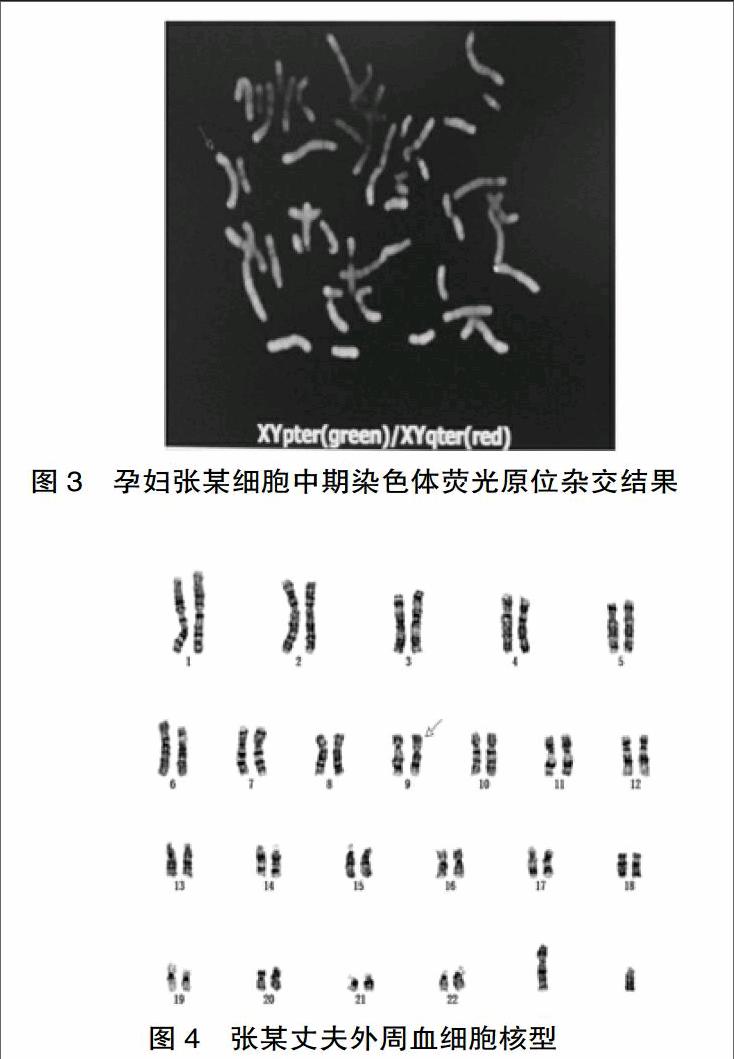
①外周血细胞G显带(320条带)染色体分析发现其一条X染色体短臂存在部分缺失(图1),由于缺失片段较小,且其本人未有明显异常表型,又进一步行SNP-array分析,以排除是否平衡性易位,SNP-array结果arr[hg19] Xp22.33p22.2(522,089-17,096,900)x1显示该患者为女性在X染色体p22.33p22.2区段存在16.57 Mb片段的缺失(图2),FISH结果: ish del(X)((p22.1)(XYpter-,XYqter+)显示患者为X染色体短臂末端缺失的携带者(图3),通过SNP-array与FISH二者相互验证,最后确定张某核型为46,X,del(X)(p22.2-ter)。②性激素六项及甲状腺功能检测结果如下:雌二醇(E2) 201 pg/mL(正常参考值99~448 pmol/L),促黄体生成素 (LH)8.62mIU/mL(正常参考值:2.12~10.89 mIU/ mL),促卵泡生成素(FSH)9.55 mIU/mL(正常参考值:4.5~22.51 mlU/mL),泌乳素(PRL)11 ng/mL(正常参考值:2.74~19.64 ng/mL),孕酮(P)2.05 nmol/L (正常参考值:0.99~4.83 nmol/L),睾酮(T)0.42 ng/mL(正常参考值:0.1~0.9 ng/mL)。游离三碘甲状腺原氨酸4.18 pmol/L(正常参考值:3.8~6.0 pmol/L),游离四碘甲状腺原氨酸 9.96 pmol/L(正常参考值:7.5~21.1 pmol/L),促甲狀腺激素4.22 ulU/m1(正常参考值:0.34~5.60 ulU/m1),甲状腺过氧化物酶抗体1 IU/mL(正常参考值:0~9 IU/mL)。
2.2 丈夫
外周血细胞G显带(320条带)染色体分析发现其一条9号染色体存在臂间倒位(图4),结合C带实验确认其核型为46,XY,inv(9)(p11q13)。
2.3 胎儿
羊水细胞G显带(320条带)染色体分析其核型为46,XX。
3 讨论
正常的女性个体应具有两条结构完整的X染色体,当X染色体发生数目或结构异常时,将会导致性腺发育不良或功能障碍,出现闭经、不孕等。X染色体的结构异常包括:缺失、易位、倒位以及等臂X染色体等。
X染色体上不同片段的缺失,可有不同的表型,短臂缺失可导致性腺退化,临床表现有幼稚外生殖器,原发闭经和不孕。长臂缺失可导致原发闭经或卵巢早衰;X染色体上相同的片段或基因缺失其临床表型也不一定相同[1-2],存在于Xp末端假常染色体区的SHOX基因突变/缺失与常染色体显性遗传的Leri-Weill软骨骨生成障碍(Leri-Weill dyschondrosteosis)等疾病相关,可致身材矮小,并且与骨骼异常如高腭弓、小颌畸形、肘外翻、短掌骨、脊柱侧凸等有关[3-5],但SHOX基因缺失导致的临床表型差异很大[6]。该文病例基因芯片分析显示该患者在X染色体p22.33p22.2区段存在16.57Mb片段的缺失,内含SHOX等78个OMIM基因,但该患者身高尚可,四肢骨骼发育正常,性激素六项及甲状腺功能检查正常,性发育和月经周期正常,这与以往的多数报道不一致,有分析显示,Turner综合征患者许多都有除45,X单体以外的细胞系[7],由于患者拒绝本人进一步检查及其父母接受染色体核型分析,无法得知患者的异常染色体是由于父母遗传或者是新生突变所致,以及患者身上是否还存在X短臂缺失以外的细胞系。
染色体倒位以9号最为常见,其人群中发生率约为1.0%~3.0%[8]。因无遗传物质丢失,9号倒位携带者一般无异常表型,但在减数分裂形成生殖细胞时,根据在配子形成中同源染色体相互配对和在倒位圈内奇数交换,臂间倒位在理论上将形成4种配子,有2种为部分重复和缺失,这些不平衡的配子可导致不孕不育、流产等生殖异常。
该研究中夫妇二人均为染色体异常携带者,其正常生育机会远小于双方染色体正常或只有一方为染色体异常携带者,故该研究对其胎儿做了产前诊断,结果显示核型正常。随访:孕40周顺产一女婴,女婴发育及表型未见异常。可见此类夫妇能生育正常后代,但再次怀孕时必须做产前诊断。
[参考文献]
[1] 马金元, 谢若翔, 范建华,等.463例Turner综合征的临床资料与染色体核型分析[J].中国优生与遗传杂志, 2013,33(11):40-41.
[2] 昌业伟, 贾庆华, 唐瑜,等.16例Turner综合征的细胞遗传学分析[J]. 中国优生与遗传杂志, 2013,33(4):62-63.
[3] Seki A, Jinno T, Suzuki E, et al. Skeletal Deformity Associated with SHOXDeficiency[J]. Clinical Pediatric Endocrinology, 2014, 23(3):65-72.
[4] Oliveira CS, Alves C. The role of the SHOX gene in the pathophysiology of Turner syndrome[J]. Endocrinología y nutrición : órgano de la Sociedad Espa ola de Endocrinología y Nutrición, 2011, 58(8):433.
[5] Child CJ, Kalifa G, Jones C, et al. Radiological Features in Patients with Short Stature Homeobox-Containing (SHOX) Gene Deficiency and Turner Syndrome before and after 2 Years of GH Treatment[J].Horm Res Paediatr,2015,84(1):14-25.
[6] Cho SY, Ki CS, Jang JH, et al. Familial Xp22.33-Xp22.12 deletion delineated by chromosomal microarray analysis causes proportionate short stature[J].American Journal of Medical Genetics Part A, 2012, 158A(6):1462.
[7] Hook EB, Warburton D. Turner syndrome revisited: review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss[J]. Human Genetics, 2014, 133(4):417.
[8] Dana M,Stoian V.Association of pericentric inversion of chromosome 9 and infertility in romanian population[J]. Mdica, 2012, 7(1):25-29.
(收稿日期:2017-01-18)
