Neuroprotective effects of Activin A on endoplasmic reticulum stress-mediated apoptotic and autophagic PC12 cell death
2017-06-05LongxingXueHongyuLiuYangCuiYueDongJiaoqiWangQiuyeJiJintingHeMinYaoYingyingWangYankunShaoJingMangZhongxinXu
Long-xing Xue, Hong-yu Liu, Yang Cui, Yue Dong, Jiao-qi Wang, Qiu-ye Ji, Jin-ting He, Min Yao, Ying-ying Wang, Yan-kun Shao, Jing Mang,, Zhong-xin Xu,
1 Department of Neurology, China-Japan Union Hospital, Jilin University, Changchun, Jilin Province, China
2 Research Center, China-Japan Union Hospital, Jilin University, Changchun, Jilin Province, China
Neuroprotective effects of Activin A on endoplasmic reticulum stress-mediated apoptotic and autophagic PC12 cell death
Long-xing Xue1, Hong-yu Liu1, Yang Cui1, Yue Dong1, Jiao-qi Wang1, Qiu-ye Ji2, Jin-ting He1, Min Yao1, Ying-ying Wang1, Yan-kun Shao1, Jing Mang1,*, Zhong-xin Xu1,*
1 Department of Neurology, China-Japan Union Hospital, Jilin University, Changchun, Jilin Province, China
2 Research Center, China-Japan Union Hospital, Jilin University, Changchun, Jilin Province, China
How to cite this article:Xue LX, Liu HY, Cui Y, Dong Y, Wang JQ, Ji QY, He JT, Yao M, Wang YY, Shao YK, Mang J, Xu ZX (2017) Neuroprotective effects of Activin A on endoplasmic reticulum stress-mediated apoptotic and autophagic PC12 cell death. Neural Regen Res 12(5):779-786.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Funding:This study was supported by the National Natural Science Foundation of China, No. 81671159, No. 81371298; a grant from the Development of Science and Technology of Jilin Province of China, No. 20160101099JC, No. 20160101073JC; a grant from the Youth Scientific Research of Health and Family Planning Commission in Jilin Province of China, No. 2014Q022; a grant from the Frontier Interdiscipline Program of Norman Bethune Health Science Center of Jilin University of China, No. 2013107028; a grant from the Young Scholars Program of Norman Bethune Health Science Center of Jilin University of China, No. 2013207052.
Graphical Abstract
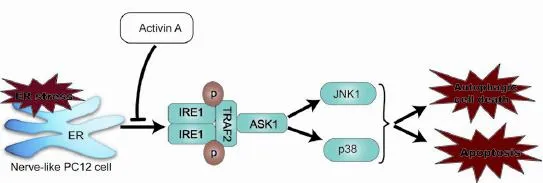
Activin A attenuates endoplasmic reticulum (ER) stress-induced apoptosis and autophagy of PC12 cells
Activin A, a member of the transforming growth factor-beta superfamily, plays a neuroprotective role in multiple neurological diseases. Endoplasmic reticulum (ER) stress-mediated apoptotic and autophagic cell death is implicated in a wide range of diseases, including cerebral ischemia and neurodegenerative diseases. Thapsigargin was used to induce PC12 cell death, and Activin A was used for intervention. Our results showed that Activin A significantly inhibited morphological changes in thapsigargin-induced apoptotic cells, and the expression of apoptosis-associated proteins [cleaved-caspase-12, C/EBP homologous protein (CHOP) and cleaved-caspase-3] and biomarkers of autophagy (Beclin-1 and light chain 3), and downregulated the expression of thapsigargin-induced ER stress-associated proteins [inositol requiring enzyme-1 (IRE1), tumor necrosis factor receptor-associated factor 2 (TRAF2), apoptosis signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinase (JNK) and p38]. The inhibition of thapsigargin-induced cell death was concentration-dependent. These findings suggest that administration of Activin A protects PC12 cells against ER stress-mediated apoptotic and autophagic cell death by inhibiting the activation of the IRE1-TRAF2-ASK1-JNK/p38 cascade.
nerve regeneration; Activin A; endoplasmic reticulum stress; apoptosis; autophagy; c-Jun N-terminal kinase; p38; neural regeneration
Introduction
The endoplasmic reticulum (ER) is a eukaryotic organelle that is the site of folding of membrane and secreted proteins, synthesis of lipids and sterols, and storage of free calcium (Lin et al., 2008; Chen et al., 2016). Cellular stress conditions, such as glucose deprivation and depletion of ER Ca2+stores, may perturb ER function, resulting in accumulation of misfolded proteins in the ER lumen, a condition referred to as ER stress. To counteract ER stress, the cells have developed a self-protective signal transduction pathway termed the unfolded protein response, which relieves cells from stress by clearing the misfolded proteins from the ER lumen. None-theless, if the ER stress is too severe, the unfolded protein response activates unique pathways that lead to cell death through apoptosis (Szegezdi et al., 2006) and autophagic cell death (Momoi, 2006). ER stress has been shown to act as the key signal for the two programmed cell death pathways (Moretti et al., 2007). As ER stress-mediated apoptotic and autophagic cell death is implicated in the pathophysiology of cerebral ischemia and a variety of neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease (Momoi, 2006; Kim et al., 2015), many studies have focused on identifying approaches to attenuate ER stress-mediated cell death in neurons.
Inositol requiring enzyme-1 (IRE1), an important ER transmembrane stress sensor, can initiate cell death in response to ER stress (Lin et al., 2008). After activation by ER stress, IRE1 binds to tumor necrosis factor-receptor-associated factor 2 (TRAF2), and then recruits and activates apoptosis signal-regulating kinase 1 (ASK1), a mitogen-activated protein (MAP) kinase kinase kinase (MAP3K). This IRE1-TRAF2-ASK1 complex promotes activation of p38 and c-Jun N-terminal kinase (JNK) during ER stress (Xin et al., 2014). JNK and p38 are two apoptosis-regulating MAP kinases (MAPK), which are activated in response to a variety of cell stresses, including reactive oxygen species, DNA damage and inflammatory-induced cytokines (Nozaki et al., 2001). A recent study has reported that the JNK and p38 MAPK cascades are involved in ER stress-induced apoptosis in some model systems of neurodegenerative diseases (Sekine et al., 2006). In addition, some studies have shown that the JNK and p38 MAPK cascades participate in autophagy activation during cerebral ischemia and neuronal degeneration (Gomez-Santos et al., 2003; Xue et al., 2016).
Previously, we have found that Activin A administration simultaneously suppressed both JNK and p38 activation after cerebral ischemia (Xue et al., 2016). Activin A, a member of the transforming growth factor-beta family, is an endogenous neuronal survival factor that is induced after various forms of acute brain disorders and injuries, including epilepsy, stroke and trauma (Brackmann et al., 2013). A growing body of evidence suggests that Activin A exerts its neuroprotective roleviasuppression of the mitochondria/cytochrome c-mediated apoptotic pathway (Mukerji et al., 2009; He et al., 2011). However, the effects of Activin A on ER stress-mediated apoptotic and autophagic cell death remain unclear. In this study, we established a model of ER stress in PC12 cells, to investigate the role of Activin A in ER stress-mediated cell death.
Materials and Methods
Culture and differentiation of PC12 cells
PC12 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Beijing Gold Amethyst Pharm & Bio-Tech Co. Ltd., Beijing, China). The cell line was grown in complete Dulbecco’s modified Eagle’s medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco) in a 5% CO2incubator at 37°C. After attachment, cells were incubated in medium containing nerve growth factor (Sigma, New York, NY, USA) at a final concentration of 100 ng/mL for 6 days, to generate nerve-like PC12 cells. Unless mentioned otherwise, all experiments were performed with nerve-like PC12 cells treated as described above.
Drug treatment
Thapsigargin (TG) is a representative ER stress inducer (Zou et al., 2015). To induce ER stress, PC12 cells were treated for 20 hours with 1 μM TG (Sigma), which was dissolved in dimethyl sulfoxide (DMSO) (Nakajima et al., 2014). PC12 cells treated with TG were the TG group. Cells with the same amount of DMSO addition were the DMSO group.
PC12 cells were pretreated with 30, 50 or 100 ng/mL Activin A (Sigma) for 24 hours, followed by co-treatment with 1 μM TG. Cells were also pretreated with specific inhibitors of p38 (SB203580, Beyotime, Shanghai, China; 10 μM) or JNK (SP600125, Beyotime; 10 μM) for 30 minutes, followed by co-treatment with 1 μM TG.
3-Methyladenine (3-MA) is a specific inhibitor of autophagy (Seglen and Gordon, 1982). Cells were pretreated with 1 mM 3-MA (Sigma) for 30 minutes, followed by co-treatment with 1 μM TG.
Cell viability assay
Cell viability was measured by the Cell Counting Kit-8 (CCK-8) assay. Briefly, PC12 cells were plated in 96-well plates and differentiated into neurons. After different treatments, the medium was replaced with 100 μL of normal medium, and 10 μL of CCK-8 solution (Dojindo Laboratories, Kumamoto, Japan) were added to each well and incubated for 2 hours. The optical density (OD) of each well was measured at 450 nm using a Universal Microplate reader (Bio-TEK Instrument. Inc., Winooski, VT, USA). The experiment was repeated three times. The cell viability rate (%) was equal to ODexperimentalgroup/ODcontrolgroup× 100%.
Acridine orange (AO) staining
AO staining was used to quantify the induction of autophagy in PC12 cells. Following treatment, the cells were incubated with 1 μg/mL AO solution (Sigma-Aldrich, St. Louis, MO, USA) for 15 minutes at room temperature in the dark. After being washed by phosphate-buffered saline (PBS), cells were visualized by fluorescence microscopy (Olympus BX61, Tokyo, Japan).
Hoechst 33342 fluorescence staining
Cells were plated in 24-well plates and induced by nerve growth factor to differentiate into neurons, and then treated according to the experimental design the next day. After the treatment, cell media were removed and 0.5 mL of Hoechst 33342 staining solution (5 μg/mL; Invitrogen, Carlsbad, CA, USA) was added to the cells. The cells were incubated in a 5% CO2incubator at 37°C for 20 minutes and rinsed with PBS twice before fluorescence microscopy analysis. Morphologic changes such as condensed or fragmented nuclei were considered to be apoptotic.
Western blot assay
Samples containing equal amounts of protein were electrophoresed in sodium dodecyl sulphate-polyacrylamide gels, and then transferred to polyvinylidene fluoride membranes. After blocking in 5% nonfat milk in Tris-buffered saline and Tween 20 (TBST) for 1 hour at room temperature, membranes were incubated with the primary antibodies in TBST overnight at 4°C. The following primary antibodies were used: mouse anti-β-actin (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-JNK1 (1:1,000; CST, Danvers, MA, USA), rabbit anti-p-JNK1 (1:1,000; CST), rabbit anti-p38 (1:1,000; ThermoFisher Scientific, Waltham, MA, USA), rabbit anti-p-p38 (1:1,000; ThermoFisher Scientific), rabbit anti-light chain 3 (LC3) (1:1,000; Abcam, Cambridge, UK), mouse anti-Beclin-1 (1:500; Santa Cruz Biotechnology), rabbit anti-cleaved caspase-3 (1:1,000; CST), rabbit anti-cleaved caspase-12 (1:1,000; Abcam), mouse anti-C/EBP homologous protein (CHOP; 1:1,000; Santa Cruz Biotechnology), goat anti-IRE1 (1:1,000; Abcam), rabbit anti-TRAF2 (1:1,000; CST) and rabbit anti-ASK1 (1:1,000; CST). The membranes were washed three times with TBST, and then probed with secondary horseradish peroxidase-conjugated anti-rabbit, anti-mouse, or anti-goat IgG (1:3,000; KPL, Gaithersburg, MD, USA) in TBST for 1 hour at room temperature. Protein densitometry was normalized against β-actin and analyzed by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Analyses were conducted using SPSS version 17.0 (SPSS, Chicago, IL, USA). All data are presented as the mean ± standard deviation (SD) of at least three independent experiments. Statistical analyses were first performed using oneway analysis of variance to determine the statistically significant variation among groups. Multiple comparisons were then evaluated using Bonferronipost hoctests. A difference was considered significant atP< 0.05.
Results
Activin A protected PC12 cells against TG-induced cell death
To investigate the effects of Activin A on ER stress-induced cell death in nerve-like PC12 cells, cells were preincubated with 30, 50 or 100 ng/mL Activin A for 24 hours, and then co-treated with the ER stress inducer, 1 μM TG, for 20 hours. Cell viability was then measured by CCK-8 assay. As shown in Figure 1, the cell viability was significantly lower in the TG group compared with the DMSO group, which was improved by Activin A in a concentration-dependent manner. One hundred nanograms per milliliter Activin A was used in the subsequent experiments.
Activin A inhibited ER stress-induced apoptotic cell death in PC12 cells
Next, we investigated whether Activin A prevents TG-induced apoptosis in PC12 cells. The nuclear morphological changes were determined by Hoechst33342 fluorescence staining. TG-treated cells showed highly condensed and fragmented nuclei, which is characteristic of apoptosis. In contrast, Activin A significantly inhibited the TG-induced apoptotic features (Figure 2A).
CHOP and caspase-12 are hallmarks of ER stress, which are involved in ER stress-dependent apoptosis (Xin et al., 2014). Caspase-3 is an apoptosis-associated protein (Li et al., 2017). The protein levels of CHOP, cleaved-caspase-12 and cleaved-caspase-3 were investigated by the western blot assay. Our results showed that TG treatment markedly increased the expression of CHOP, cleaved-caspase-12 and cleaved-caspase-3, but the alterations were significantly inhibited by Activin A treatment (Figure 2B).
Activin A inhibited ER stress-induced autophagic cell death in PC12 cells
To test the effects of exogenous Activin A on autophagy activation following ER stress, the expression of microtubule-associated protein 1 light chain 3 (LC3-II) and Beclin-1, two autophagy indicators (Shi et al., 2012), was examined. As shown in Figure 3A, the relative expression of both Beclin-1 and LC3-II significantly increased following TG treatment, and Activin A significantly reduced this elevated expression. AO is a fluorescent substance that is often used to detect autophagy (Luo et al., 2014). As shown in Figure 3D, compared with the DMSO group, TG treatment induced significantly more red AO spots in the cytoplasm of PC12 cells. However, the effect was suppressed by treatment with Activin A.
To determine the role of autophagy in ER stress-induced neuronal injury, PC12 cells were treated with the autophagy inhibitor, 3-MA, during ER stress. Treatment with 1 mM 3-MA effectively blocked the ER stress-induced autophagy as evidenced by inhibiting the production of LC3-II and Beclin-1 (Figure 3B), whereas 3-MA treatment alone had no effect on the expression of LC3-II and Beclin-1. Furthermore, we found that 3-MA reduced the ER stress-induced cell death significantly, while treatment with 1 mM 3-MA alone had no effect on cell viability (Figure 3C).
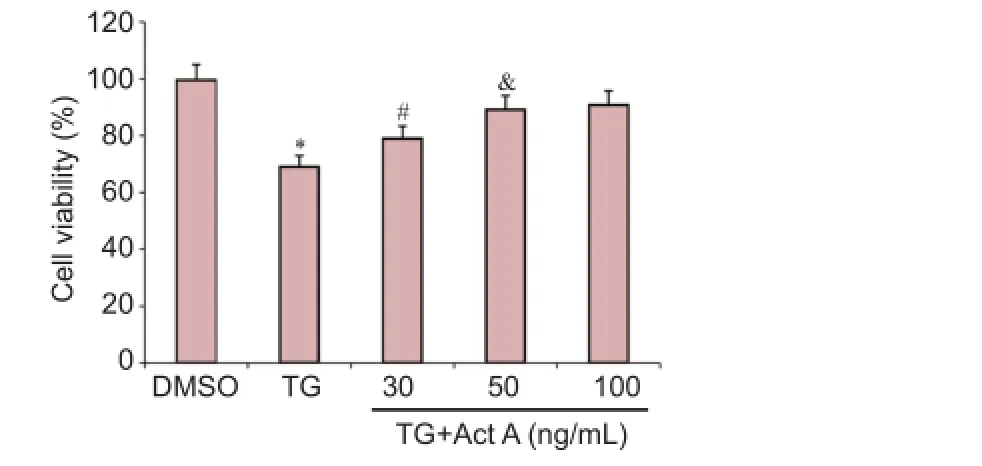
Figure 1 Act A protected PC12 cells against endoplasmic reticulum stress-induced cell death.
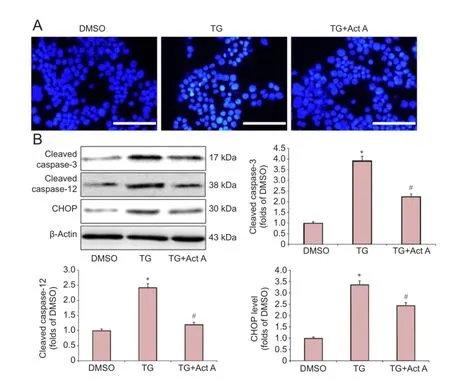
Figure 2 Act A reduced endoplasmic reticulum stress-induced apoptosis in PC12 cells.
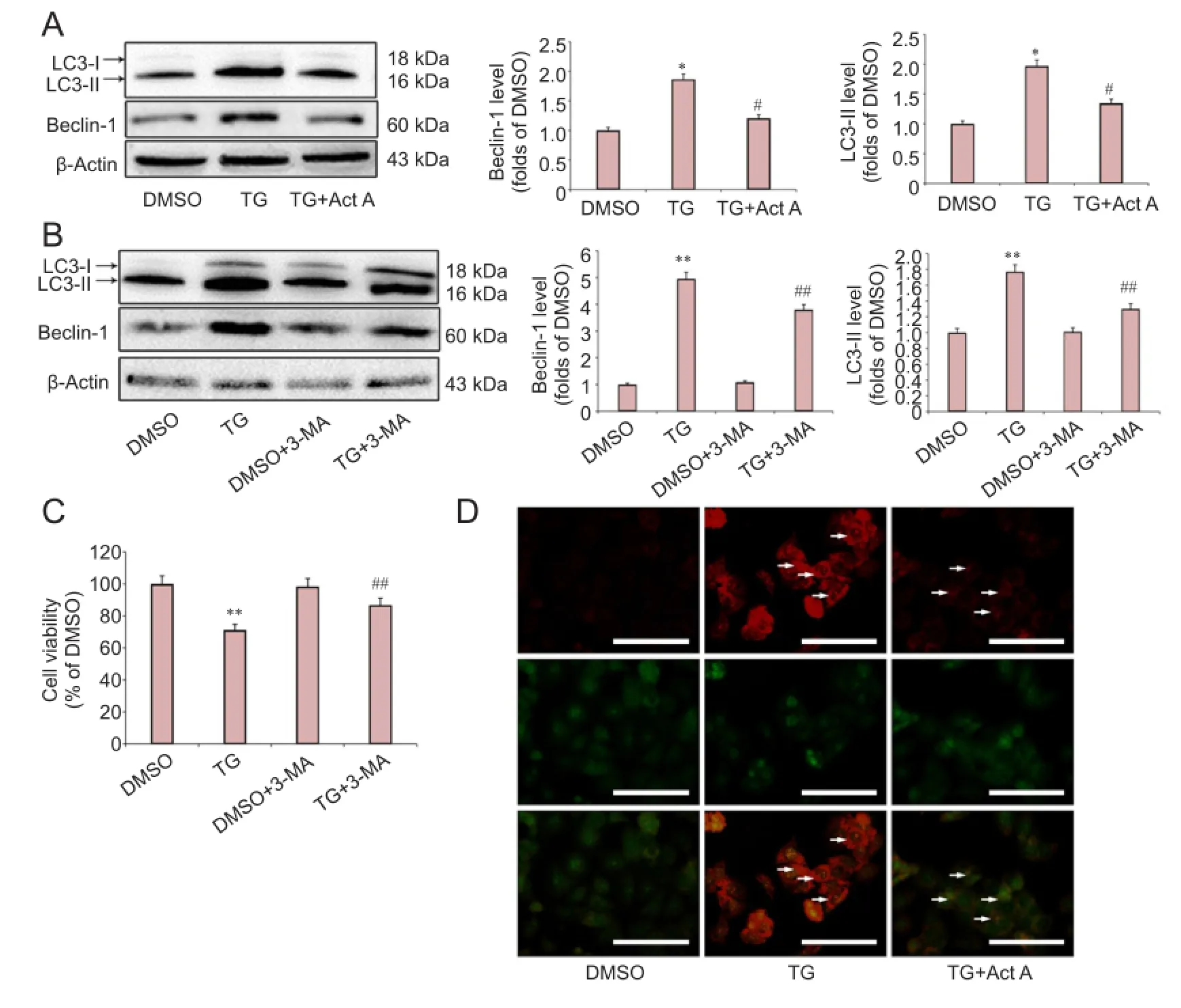
Figure 3 Act A reduced endoplasmic reticulum stress-induced autophagic cell death in PC12 cells.
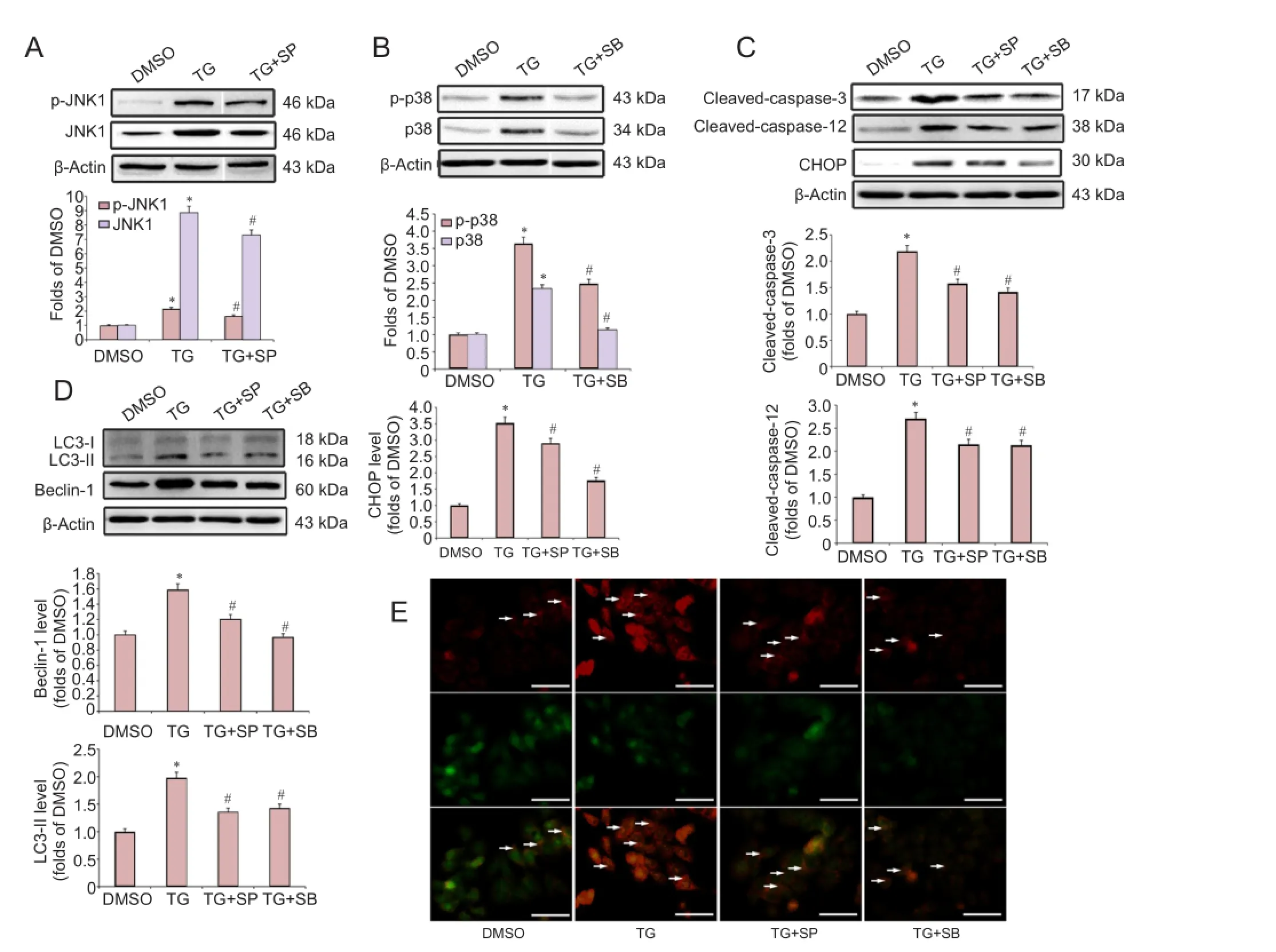
Figure 4 The JNK and p38 MAPK pathways were involved in the endoplasmic reticulum stress-induced apoptosis and autophagy in PC12 cells.
The JNK and p38 MAPK pathways were involved in ER stress-induced apoptosis and autophagy in PC12 cells
Recent studies have reported that the JNK and p38 MAPK pathways are involved in apoptosis and autophagy activation during cerebral ischemia and neuronal degeneration (Gomez-Santos et al., 2003; Sekine et al., 2006; Xue et al., 2016). To determine the role of JNK and p38 MAPK signaling in the ER stress-induced apoptosis, cells were pretreated with specific inhibitors of p38 (10 μM SB203580) and JNK (10 μM SP600125), followed by co-treatment with TG for 20 hours. Western blot analysis showed that both JNK and p38 were activated by TG treatment, and pretreatment with SP600125 or SB203580 directly inhibited the TG-induced activation of JNK and p38 MAPK, respectively (Figure 4A, B). Furthermore, our results showed that both SP600125 and SB203580 significantly attenuated the TG-induced elevation in the protein levels of CHOP, cleaved-caspase-12 and cleaved-caspase-3 (Figure 4C).
We then investigated the role of the JNK and p38 MAPK pathways in ER stress-induced autophagy. As shown in Figure 4D, pretreatment with either SB203580 or SP600125 markedly attenuated the TG-induced elevation in the protein levels of Beclin-1 and LC3-II. Moreover, SB203580 and SP600125 treatments suppressed the TG-induced strong AO punctate fluorescence in the cytoplasm (Figure 4E).
Activin A suppressed ER stress-induced activation of the IRE1-TRAF2-ASK1-JNK/p38 cascade
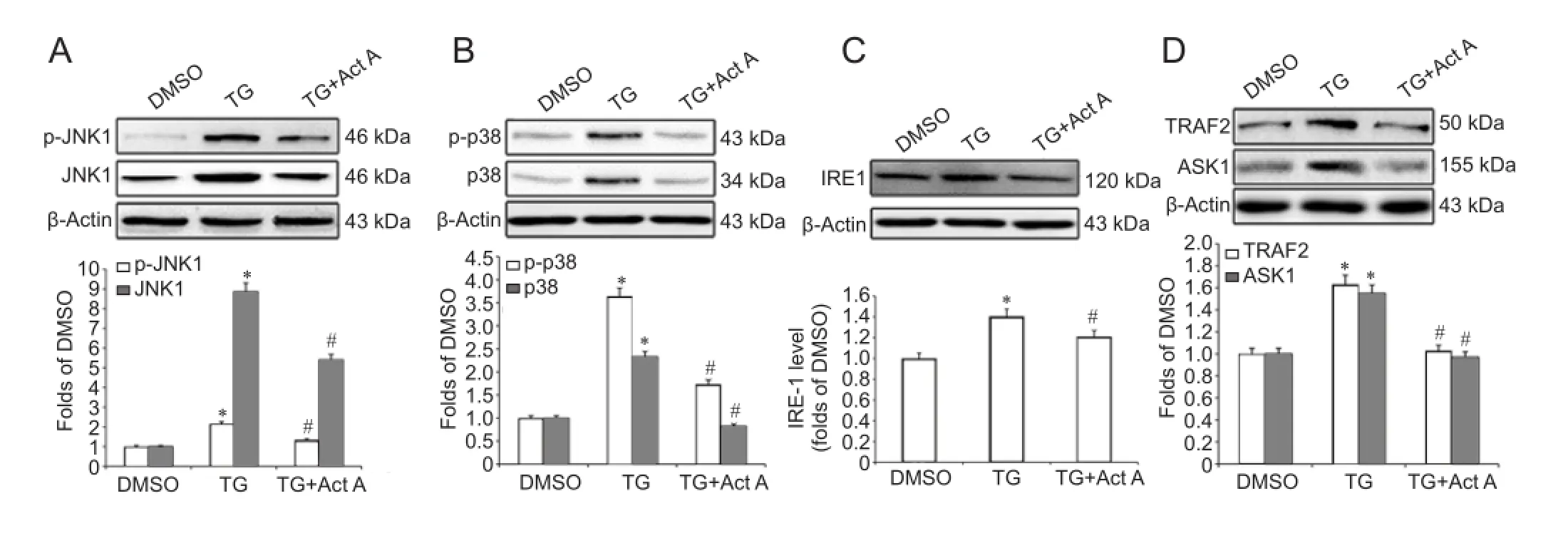
Figure 5 Act A (100 ng/mL) suppressed the endoplasmic reticulum stress-induced IRE1-TRAF2-ASK1-JNK/p38 cascade in PC12 cells.
To investigate the potential mechanisms by which Activin A inhibits ER stress-induced apoptosis and autophagy, we examined the effects of exogenous Activin A on the JNK and p38 MAPK pathways during ER stress. We found that the TG-induced elevation in the protein levels of JNK, p-JNK, p38 and p-p38 was significantly inhibited by Activin A (Figure 5A, B).
Studies have shown that JNK and p38 MAPK are the downstream effectors of the IRE1-TRAF2-ASK1 pathway during ER stress (Xin et al., 2014; Zou et al., 2015). ASK1 is a member of the large MAP3K family that activates both the JNK and p38 MAPK pathways (Xin et al., 2014). To further investigate the potential mechanisms by which exogenous Activin A suppresses JNK/p38 activation during ER stress, we examined the protein levels of IRE1, TRAF2 and ASK1 in TG and Activin A-treated PC12 cells. Western blot assay revealed that IRE1, TRAF2 and ASK1 were upregulated in the TG group, while Activin A significantly attenuated this upregulation (Figure 5C, D).
Discussion
ER stress-mediated apoptotic and autopahgic cell death has been implicated in many diseases, including cerebral ischemia (Lin, 2015), secondary brain damage (Sun et al., 2016), and a variety of neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease (Momoi, 2006; Kim et al., 2015). Many studies have focused on identifying approaches to attenuate ER stress-mediated cell death in neurons. Activin A, a member of the transforming growth factor-beta superfamily, plays a neuroprotective role in multiple neurological diseases. In this study, we found that Activin A attenuated PC12 cell death induced by the representative ER stress inducer, TG, in a concentration-dependent manner. The highly differentiated rat pheochromocytoma (PC12) cells share many typical properties with neuronal cells, including outgrowth of neurites, synthesis of neurotransmitters, selective expression of certain proteins and interactions of compounds with membrane bound receptors (Shafer and Atchison, 1991). We thus used the PC12 cell lines to establish ER stress model in the present study.
CHOP and caspase-12 are two hallmarks of ER stress, which are involved in ER stress-dependent apoptosis (Xin et al., 2014). Prolonged elevation in the expression of the pro-apoptotic transcription factor, CHOP, triggers apoptosis by affecting the intracellular calcium metabolism and by alterations in Bcl family members (Timmins et al., 2009). Caspase-12, an ER-specific caspase, is normally in the inactive pro-caspase form (Nakagawa et al., 2000). During ER stress, caspase-12 dissociates from the ER membrane and is cleaved, resulting in its activation, which leads directly or indirectly to the cleavage of caspase-3 (Zhao et al., 2013). In this study, we found that the protein levels of CHOP, cleavedcaspase-12 and cleaved-caspase-3 were greatly upregulated after TG treatment, whereas Activin A significantly reduced the expression of these proteins. Furthermore, Activin A significantly suppressed TG-induced apoptosis as demonstrated by Hoechst 33342 staining. These results demonstrated that exogenous Activin A attenuated ER stress-induced apoptosis in PC12 cells.
Mounting evidence suggests that ER stress is a potent trigger of autophagy, a process whereby eukaryotic cells recycle their macromolecules and organelles (Høyer-Hansen and Jäättelä, 2007). Depending on the extent of ER stress, autophagy enhances cell survival through degradation of misfolded proteins that have been retained in the ER (Qin et al., 2003), or commits the cells to autophagic cell death through excessive self-digestion and degradation of essential cellular constituents (Wen et al., 2008). Because of the high activity and reliance on autophagy in neurons, these cells may be particularly susceptible to autophagic cell death (Button et al., 2015). Autophagic cell death has been observed in the brains of patients with cerebral ischemia (Wen et al., 2008), and neurodegenerative diseases such as Huntington’s disease and Parkinson’s disease (Kegel et al.,2000; Stefanis et al., 2001). In this study, we found that the expression of LC3-II and Beclin-1, two autophagy indicators, increased following TG treatment. LC3 is found in the cytosolic form, LC3-I, and the membrane bound form, LC3-II. During autophagy maturation, LC3-I converts to LC3-II after cleavage of a few amino acids at the C-terminus, hence autophagic activity, defined in terms of autophagosome amount, directly correlates with the LC3-II amount or with the ratio of LC3-II/LC3-I (Sheng et al., 2012). Beclin-1 is essential for the recruitment of other autophagic proteins during the expansion of pre-autophagosomal membranes (Zhang et al., 2015). Our results suggest that autophagy was activated during ER stress. Furthermore, we found that inhibition of autophagy by 3-MA effectively decreased the TG-induced cell death. These data demonstrated that autophagy was involved in the cell death induced by ER stress in PC12 cells.
To test the effects of exogenous Activin A on autophagy activation following ER stress, cells were pretreated with exogenous Activin A, followed by co-treatment with TG for 20 hours. We found that Activin A significantly suppressed the TG-induced increase in the expression of LC3-II and Beclin-1 in PC12 cells. These results were further confirmed by AO staining. Autophagy is characterized by the development of acidic vesicular organelles, which include lysosomes and autophagosomes (Paglin et al., 2001). AO is a nucleic acid-selective fluorescent cationic dye that stains the nucleus and cytoplasm green, and any acidic compartments red (Yang et al., 2014). In this study, we found that TG-induced red AO spots in the cytoplasm of PC12 cells were suppressed by Activin A. These results suggest that exogenous Activin A inhibited ER stress-induced autophagy in PC12 cells. Combined with the autophagy effects on ER stress-induced cell death, we concluded that exogenous Activin A protected PC12 cells against ER stress-induced injury partly by attenuating autophagic cell death.
JNK and p38 MAPK are two apoptosis-regulating MAP kinases, which are activated in response to a variety of cell stresses including reactive oxygen species, DNA damage and inflammatory-induced cytokines (Nozaki et al., 2001). Recent studies have shown that the JNK and p38 MAPK pathways are involved in apoptosis and autophagy activation during cerebral ischemia and neuronal degeneration (Gomez-Santos et al., 2003; Sekine et al., 2006; Xue et al., 2016). In this study, we found that JNK and p38 inhibition attenuated TG-induced autophagy in PC12 cells. In addition, JNK and p38 inhibition markedly attenuated the TG-induced elevation in the protein levels of CHOP, cleaved-caspase-12 and cleaved-caspase-3. These findings suggest that the JNK and p38 pathways were involved in the autophagy and apoptosis during ER stress.
Previously, we have found that Activin A simultaneously suppressed the activation of the JNK and p38 pathways during cerebral ischemia (Xue et al., 2016). In this study, we found that the TG-induced activation of the JNK/p38 pathways was significantly inhibited by exogenous Activin A. The simultaneous reduction in both p38 and JNK activation indicated that exogenous Activin A may regulate a common upstream kinase during ER stress. Several studies have shown that JNK and p38 MAPK are the downstream effectors of the IRE1-TRAF2-ASK1 pathway during ER stress (Xin et al., 2014; Zou et al., 2015). In response to ER stress, IRE1 binds to TRAF2, which then recruits and activates ASK, a MAP3K. This IRE1-TRAF2-ASK1 complex promotes activation of p38 and JNK during ER stress (Xin et al., 2014). In this study, Activin A inhibited the TG-induced expression of IRE1, TRAF2 and ASK1 in PC12 cells. These results suggest that Activin A inhibited the activation of the IRE1-TRAF2-ASK1-JNK/p38 cascade during ER stress.
In summary, Activin A inhibited ER stress-induced apoptotic and autophagic cell death by suppressing the IRE1-TRAF2-ASK1-JNK/p38 cascade in PC12 cells. Because ER stress has increasingly come into focus as a factor contributing to neuronal injury, further understanding of the protective effects of Activin A against ER stress may significantly enhance its pharmaceutical potential for treatment of neurological diseases.
Author contributions:JM, ZXX and LXX designed experiments. LXX, HYL, YD and JQW carried out experiments. YC and JTH analyzed experimental results. LXX wrote the paper. QYJ, YYW, MY and YKS assisted with experiments and paper writing. All authors approved the final version of the paper.
Conflicts of interest:None declared.
Plagiarism check:This paper was screened twice using CrossCheck to verify originality before publication.
Peer review:This paper was double-blinded and stringently reviewed by international expert reviewers.
Brackmann FA, Link AS, Jung S, Richter M, Zoglauer D, Walkinshaw G, Alzheimer C, Trollmann R (2013) Activin A regulation under global hypoxia in developing mouse brain. Brain Res 1531:65-74.
Button RW, Luo S, Rubinsztein DC (2015) Autophagic activity in neuronal cell death. Neurosci Bull 31:382-394.
Chen ZK, Li XF, Zhang HN (2016) Effects of cyclic tensile stress on caspase-12 expression in chondrocytes. Zhongguo Zuzhi Gongcheng Yanjiu 20:4334-4340.
Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S (2003) Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res 73:341-350.
He JT, Mang J, Mei CL, Yang L, Wang JQ, Xing Y, Yang H, Xu ZX (2011) Neuroprotective effects of exogenous activin A on oxygen-glucose deprivation in PC12 cells. Molecules 17:315-327.
Høyer-Hansen M, Jäättelä M (2007) Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 14:1576-1582.
Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M (2000) Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci 20:7268-7278.
Kim J, Moon IS, Goo TW, Moon SS, Seo M (2015) Algae undaria pinnatifida protects hypothalamic neurons against endoplasmic reticulum stress through Akt/mTOR signaling. Molecules 20:20998-21009.
Li Z, Wu F, Zhang X, Chai Y, Chen D, Yang Y, Xu K, Yin J, Li R, Shi H, Wang Z, Li X, Xiao J, Zhang H (2017) Valproate attenuates endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells via the AKT/GSK3beta signaling pathway. Int J Mol Sci 18.
Lin CL (2015) Attenuation of endoplasmic reticulum stress as a treatment strategy against ischemia/reperfusion injury. Neural Regen Res 10:1930-1931.
Lin JH, Walter P, Yen TS (2008) Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 3:399-425.
Luo T, Liu G, Ma H, Lu B, Xu H, Wang Y, Wu J, Ge P, Liang J (2014) Inhibition of autophagy via activation of PI3K/Akt pathway contributes to the protection of ginsenoside Rb1 against neuronal death caused by ischemic insults. Int J Mol Sci 15:15426-15442.
Momoi T (2006) Conformational diseases and ER stress-mediated cell death: apoptotic cell death and autophagic cell death. Curr Mol Med 6:111-118.
Moretti L, Cha YI, Niermann KJ, Lu B (2007) Switch between apoptosis and autophagy: radiation-induced endoplasmic reticulum stress? Cell Cycle 6:793-798.
Mukerji SS, Rainey RN, Rhodes JL, Hall AK (2009) Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J Neurochem 111:1138-1148.
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403:98-103.
Nakajima A, Tsuji M, Inagaki M, Tamura Y, Kato M, Niiya A, Usui Y, Oguchi K (2014) Neuroprotective effects of propofol on ER stress-mediated apoptosis in neuroblastoma SH-SY5Y cells. Eur J Pharmacol 725:47-54.
Nozaki K, Nishimura M, Hashimoto N (2001) Mitogen-activated protein kinases and cerebral ischemia. Mol Neurobiol 23:1-19.
Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61:439-444.
Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M (2003) Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet 12:3231-3244.
Seglen PO, Gordon PB (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 79:1889-1892.
Sekine Y, Takeda K, Ichijo H (2006) The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Curr Mol Med 6:87-97.
Shafer TJ, Atchison WD (1991) Transmitter, ion channel and receptor properties of pheochromocytoma (PC12) cells: a model for neurotoxicological studies. Neurotoxicology 12:473-492.
Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, Zhang XY, Qin ZH (2012) Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy 8:310-325.
Shi R, Weng J, Zhao L, Li XM, Gao TM, Kong J (2012) Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther 18:250-260.
Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA (2001) Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 21:9549-9560.
Sun GZ, Gao FF, Zhao ZM, Sun H, Xu W, Wu LW, He YC (2016) Endoplasmic reticulum stress-induced apoptosis in the penumbra aggravates secondary damage in rats with traumatic brain injury. Neural Regen Res 11:1260-1266.
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7:880-885.
Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I (2009) Calcium/ calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest 119:2925-2941.
Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH (2008) Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 4:762-769.
Xin Q, Ji B, Cheng B, Wang C, Liu H, Chen X, Chen J, Bai B (2014) Endoplasmic reticulum stress in cerebral ischemia. Neurochem Int 68:18-27.
Xue LX, Xu ZH, Wang JQ, Cui Y, Liu HY, Liang WZ, Ji QY, He JT, Shao YK, Mang J, Xu ZX (2016) Activin A/Smads signaling pathway negatively regulates oxygen glucose deprivation-induced autophagy via suppression of JNK and p38 MAPK pathways in neuronal PC12 cells. Biochem Biophys Res Commun 480:355-361.
Yang Z, Zhao TZ, Zou YJ, Zhang JH, Feng H (2014) Hypoxia Induces autophagic cell death through hypoxia-inducible factor 1alpha in microglia. PLoS One 9:e96509.
Zhang XY, Zhang TT, Song DD, Zhou J, Han R, Qin ZH, Sheng R (2015) Endoplasmic reticulum chaperone GRP78 is involved in autophagy activation induced by ischemic preconditioning in neural cells. Mol Brain 8:20.
Zhao S, Xiong Z, Mao X, Meng D, Lei Q, Li Y, Deng P, Chen M, Tu M, Lu X, Yang G, He G (2013) Atmospheric pressure room temperature plasma jets facilitate oxidative and nitrative stress and lead to endoplasmic reticulum stress dependent apoptosis in HepG2 cells. PLoS One 8:e73665.
Zou H, Limpert AS, Zou J, Dembo A, Lee PS, Grant D, Ardecky R, Pinkerton AB, Magnuson GK, Goldman ME, Rong J, Teriete P, Sheffler DJ, Reed JC, Cosford ND (2015) Benzodiazepinone derivatives protect against endoplasmic reticulum stress-mediated cell death in human neuronal cell lines. ACS Chem Neurosci 6:464-475.
Copyedited by Yu J, Li CH, Qiu Y, Song LP, Zhao M
*< class="emphasis_italic">Correspondence to: Jing Mang or Zhong-xin Xu, mangjing@jlu.edu.cn or xuzhongxin999@aliyun.com.
Jing Mang or Zhong-xin Xu, mangjing@jlu.edu.cn or xuzhongxin999@aliyun.com.
orcid: 0000-0003-3154-9218 (Jing Mang) 0000-0001-7066-7159 (Zhong-xin Xu)
10.4103/1673-5374.206649
Accepted: 2017-04-20
杂志排行

中国神经再生研究(英文版)的其它文章
- Cerebral mechanism of puncturing at He-Mu point combination for functional dyspepsia: study protocol for a randomized controlled parallel trial
- Therapeutic opportunities and challenges of induced pluripotent stem cells-derived motor neurons for treatment of amyotrophic lateral sclerosis and motor neuron disease
- Inhibition and enhancement of neural regeneration by chondroitin sulfate proteoglycans
- Collapsin response mediator protein-2 plays a major protective role in acute axonal degeneration
- Hypoxia inducible factor-1 alpha stabilization for regenerative therapy in traumatic brain injury
- Minocycline targets multiple secondary injury mechanisms in traumatic spinal cord injury