Minocycline targets multiple secondary injury mechanisms in traumatic spinal cord injury
2017-06-05RobertShultzYinghuiZhong
Robert B. Shultz, Yinghui Zhong
School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA
Minocycline targets multiple secondary injury mechanisms in traumatic spinal cord injury
Robert B. Shultz, Yinghui Zhong*
School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA
How to cite this article:Shultz RB, Zhong Y (2017) Minocycline targets multiple secondary injury mechanisms in traumatic spinal cord injury. Neural Regen Res 12(5):702-713.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Minocycline hydrochloride (MH), a semi-synthetic tetracycline derivative, is a clinically available antibiotic and anti-inflammatory drug that also exhibits potent neuroprotective activities. It has been shown to target multiple secondary injury mechanisms in spinal cord injury,viaits anti-inflammatory, anti-oxidant, and anti-apoptotic properties. The secondary injury mechanisms that MH can potentially target include inflammation, free radicals and oxidative stress, glutamate excitotoxicity, calcium influx, mitochondrial dysfunction, ischemia, hemorrhage, and edema. This review discusses the potential mechanisms of the multifaceted actions of MH. Its anti-inflammatory and neuroprotective effects are partially achieved through conserved mechanisms such as modulation of p38 mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/Akt signaling pathways as well as inhibition of matrix metalloproteinases (MMPs). Additionally, MH can directly inhibit calcium influx through the N-methyl-D-aspartate (NMDA) receptors, mitochondrial calcium uptake, poly(ADP-ribose) polymerase-1 (PARP-1) enzymatic activity, and iron toxicity. It can also directly scavenge free radicals. Because it can target many secondary injury mechanisms, MH treatment holds great promise for reducing tissue damage and promoting functional recovery following spinal cord injury.
minocycline; inflammation; anti-oxidant; neuroprotection; oxidative stress; glutamate exitotoxicity; cytochrome c; P38 MAPK; PI3K/Akt; calcium influx
Introduction
Traumatic spinal cord injury (SCI) causes deleterious functional loss below the level of injury. The initial trauma results in rapid hemorrhage and cell death, and offers little opportunity for therapeutic intervention. Following the initial trauma, secondary injury cascades occur, causing widespread and persistent inflammation and progressive tissue loss. During this stage, lesions can become many times larger than the initial injury (Fitch et al., 1999; Rossignol et al., 2007; Fehlings and Nguyen, 2010). Therapies that can inhibit secondary injury progression thus offer a promising and clinically viable approach to reduce tissue damage and functional deficits following SCI.
Many mechanisms contribute to the secondary injury, including inflammation, cellular damage from free radicals such as reactive oxygen species (ROS) and reactive nitrogen species (RNS), glutamate exitotoxicity, calcium influx, ischemia, hemorrhage, and edema (Oyinbo, 2011). However, most current therapies only target one or a few elements of the secondary injury mechanisms, and have been largely unsuccessful in clinical trials (Thuret et al., 2006; Lammertse, 2013; Varma et al., 2013).
Minocycline hydrochloride (MH), a semi-synthetic tetracycline derivative, is a clinically available antibiotic and anti-inflammatory drug that also exhibits potent neuroprotective activities. It can potentially target a broad range of secondary injury mechanisms, and protect neural tissue from multiple neurotoxic insults after SCI,viaits anti-inflammatory, anti-oxidant, and anti-apoptotic properties (Stirling et al., 2005; Elewa et al., 2006; Sapadin and Fleischmajer, 2006; Plane et al., 2010; Ghazali et al., 2016; Chin et al., 2017). MH has been shown to (1) inhibit inflammatory processes contributing to progression of secondary injury (Lee et al., 2003a); (2) protect neurons from oxidative stress and scavenge free radicals (Lee et al., 2003a); (3) inhibit inducible nitric oxide synthase (iNOS) that produces nitric oxide (NO) (Amin et al., 1996); (4) prevent glutamate-induced apoptosis of neurons (Pi et al., 2004); (5) prevent N-methyl-D-aspartate (NMDA)-induced excitotoxicity by diminishing NMDA-induced Ca2+influx and mitochondria Ca2+uptake (Garcia-Martinez et al., 2010); (6) prevent apoptosis by inhibiting mitochondrial cytochrome c (CytC) release after SCI (Teng et al., 2004); (7) inhibit oligodendrocyte apoptosis and improve functional recovery after SCI (Stirling et al., 2004); (8) protect grey and white matter from spinal cord ischemia (Takeda et al., 2011); (9) protect neurons from hemorrhage-induced toxicity (Takeda et al., 2011); and (10) protect blood-brain barrier and reduces edema following intracerebral hemorrhage (Wasserman and Schlichter, 2007). Thus, MH can serve as a multifaceted agent that targets multiple mechanisms contributing to secondary injury and has great therapeuticpotential for the treatment of SCI.
Although there is a wealth of evidence supporting the efficacy of MH treatment following SCI in animal models, a comprehensive discussion of the multiple mechanisms of action within this context is missing. The mechanisms of action can be classified into three categories: (1) anti-inflammatory activity; (2) anti-oxidative activity; and (3) direct neuroprotective activity. In this review, we discuss the possible mechanisms by which MH exerts these effects to reduce secondary injury after SCI.
Mechanisms of Anti-Inflammatory Activity
Inflammation is a key mediator of secondary injury progression in SCI. Following initial injury, resident microglia become activated to pro-inflammatory phenotypes, while blood-borne factors and leukocytes infiltrate the spinal cord tissue (Byrnes et al., 2006; Zhou et al., 2014). In the mileu of cellular signals that follow, a complex network of cross-talk is established among recruited peripheral leukocytes, resident microglia, and astrocytes, resulting in further upregulation of neurotoxic and pro-inflammatory cytokines and chemokines (McTigue et al., 1998; Gonzalez et al., 2003; Pineau and Lacroix, 2007; Stammers et al., 2012); increased production of cytotoxic ROS/RNS (Xu et al., 2005; Cooney et al., 2014); upregulation of regeneration-inhibitory molecules including proteoglycans and the myelin-derived inhibitors Nogo-A, myelin-associated glycoprotein (MAG), and oligodendrocyte myelin glycoprotein (OMgp) (Filbin, 2003; Schweigreiter and Bandtlow, 2006; Yiu and He, 2006; Dou et al., 2009); and formation of the inhibitory glial scar (Pekny and Nilsson, 2005; Yiu and He, 2006). While inflammation has also been shown to promote clearance of debris and regeneration following SCI (David et al., 2012), therapeutic strategies that mitigate inflammation have been shown to promote cell survival and functional recovery after SCI (Lee et al., 2003a; Stirling et al., 2004; Wang et al., 2017), probably because inflammation is excessive at least at the acute stage (Gensel and Zhang, 2015). MH has been found to modulate inflammation through a number of pathways—a detailed illustration is presented in Figure 1.
Regulation of pP38 mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/Akt inflammatory signaling pathways
Inflammation is associated with activation (phosphorylation) of p38 MAPK (Figure 1), a protein kinase with a number of pro-inflammatory downstream effects (Yang et al., 2014). Activation of p38 MAPK results in activation and/or nuclear translocation of pro-inflammatory transcription factors, including nuclear factor kappaB (NF-κB) (Olson et al., 2007), lipopolysaccharide-induced tumor necrosis factor-alpha factor (LITAF) (Ceccarelli et al., 2015), Nur77 (Pang et al., 2012), activator protein 1 (AP-1) (Slomiany and Slomiany, 2013), and activating transcription factor 2 (ATF-2) (Hirose et al., 2009). These transcription factors regulate synthesis of leukocyte-recruiting chemokines and pro-inflammatory cytokines, including monocyte-chemoattractant protein-1 (MCP-1) (Hacke et al., 2010), tumor necrosis factor α (TNFα), interleukin-1β (IL-1β) and interleukin-6 (IL-6) (Olson et al., 2007; Pang et al., 2012; Yu et al., 2014). MH has been shown to inhibit phosphorylation of p38 MAPK (Yune et al., 2007; Corsaro et al., 2009; Pang et al., 2012), reduce activation and translocation of inflammation-associated transcription factors such as NF-κB, LITAF, and Nur77 (Pang et al., 2012; Song et al., 2016), and decrease expression of pro-inflammatory cytokines and chemokines bothin vitroandin vivo(Lee et al., 2003b; Kielian et al., 2007; Cai et al., 2010; Pang et al., 2012; Switzer et al., 2012). MH treatment has also been shown to maintain activation of PI3K/Akt (Pang et al., 2012; Hahn et al., 2016), a negative regulator of p38 MAPK (Guha and Mackman, 2002). Inhibition of PI3K/Akt and subsequent stimulation of p38 MAPK was shown to ameliorate the effects of MH on transcription factor activation/translocation and cytokine expression (Pang et al., 2012), suggesting that MH regulation of transcription factors and cytokine/ chemokine expression are, at least in part, downstream effects of p38MAPK inhibition.
Inflammation-induced p38MAPK activation also leads to increased expression of iNOS (Choi et al., 2005; Sung et al., 2012) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunit (Spencer et al., 2016). NO and superoxides produced by iNOS and NADPH oxidase are highly reactive species capable of damaging cell membranes, proteins, nucleic acids, and organelles, resulting in cell death. At the same time, NO also acts as a pro-inflammatory signaling molecule (Korhonen et al., 2005; Sharma et al., 2007), while superoxides can participate in the formation of lipid-derived pro-inflammatory signaling molecules (Yadav and Ramana, 2013). MH has been shown to reduce iNOS expression and NO production from reactive microglia and macrophages (Amin et al., 1996; Zhang et al., 2014), and to prevent the overexpression of NADPH oxidase in the remission phase of experimental multiple sclerosis (MS) microglia (Di Filippo et al., 2016). No direct effect of MH was found on the enzymatic activity of iNOS, suggesting a regulatory effect on gene expression was responsible for decreases in NO production (Amin et al., 1996). It is possible that MH regulation of iNOS/NADPH oxidase expression is a downstream effect of inhibiting p38 MAPK pathway.
Inflammation-activated p38 MAPK has also been shown to regulate microglial expression of proNGF, a nerve growth factor (NGF) precursor (Yune et al., 2007). ProNGF has been shown to act as a distinct ligand, activating a death-inducing receptor complex in neurons (Nykjaer et al., 2004; Hempstead, 2009) and inducing oligodendrocyte death following SCI (Beattie et al., 2002). MH has been shown to inhibit p38 MAPK activation and microglial proNGF expression following SCI, resulting in improved oligodendrocyte survival (Yune et al., 2007). In addition, treatment with a specific p38 MAPK inhibitor reduced proNGF expression from lipopolysaccharide (LPS) stimulated microgliain vitro. This data clearly illustrates that p38 MAPK activation is a prerequisitefor inflammation-induced proNGF expression that can be targeted by MH treatment.
Regulation of phospholipase A2
Inflammation also results in upregulation and activation of a class of enzymes known as phospholipase A2s (PLA2s) that break down membrane phospholipids, yielding arachidonic acid (AA). AA is then metabolized into prostaglandins and leukotrienes by cyclooxygenase (COX) and lipoxygenase (LOX), respectively. Both prostaglandins and leukotrienes are potent pro-inflammatory mediators that are then secreted into the extracellular space (Balsinde et al., 2002). Prostaglandins and leukotrienes have been shown to exacerbate secondary injury by increasing vascular permeability and peripheral immune cell invasion following SCI (Xu et al., 1990; Sharma et al., 1993; Liu and Xu, 2010), and have been implicated in chronic neuropathic pain (Zhao et al., 2007; Buczynski et al., 2010). Treatment with a dual COX/LOX inhibitor reduced inflammation and mechanical hypersensitivity following SCI (Dulin et al., 2013).
SCI is associated with upregulation/increased activity of multiple PLA2 isoforms including cytosolic PLA2 (cPLA2) and secretory PLA2 (sPLA2) (Titsworth et al., 2008; Liu et al., 2014), cyclooxygenase-2 (COX2) (Resnick et al., 1998) and 5-lipoxygenase (5-LOX) (Genovese et al., 2005). MH has been shown to reduce cPLA2 expression following neurologic injury (Ma et al., 2010), and directly inhibit sPLA2 activity in cell-free conditions, potentiallyviabinding site interference (Pruzanski et al., 1992; Dalm et al., 2010). MH has also been shown to inhibit monocyte and microglial expression of COX2 and production of pro-inflammatory prostaglandin E2 (Krady et al., 2005; Pang et al., 2012), and suppress 5-LOX expression and activation in the injured central nervous system (Chu et al., 2007, 2010). These effects of MH treatment may be partially due to inhibition of p38 MAPK pathways, as the involvement of p38 MAPK signaling pathways in cPLA2 upregulation and activation has been well established (Waterman et al., 1996; Hernández et al., 1999; Coulon et al., 2003; Kriem et al., 2005; Nito et al., 2008). Similarly, p38 MAPK has been shown to regulate LPS-induced upregulations in COX2 expression (Chen et al., 1999; Dean et al., 1999). Upregulation of 5-LOX has been associated with increased NF-κB binding in LPS-stimulated macrophages (Altavilla et al., 2009), and activation of 5-LOX is achieved by kinases downstream of p38 MAPK (Werz et al., 2000). In addition, treatment with p38 MAPK inhibitors significantly reduced inflammation-associated expression and activation of cPLA2 (Zhu et al., 2001; Kriem et al., 2005; Nito et al., 2008), COX2 (Newton et al., 2000; Nagano et al., 2002), and 5-LOX (Boden et al., 2000; Werz et al., 2000). MH likely inhibits sPLA2 isoforms predominantlyviap38-independent direct interference with binding sites (Pruzanski et al., 1992; Dalm et al., 2010). Although associations have been made between p38 MAPK activation and sPLA2 expression/activity (Rosenson and Gelb, 2009), further investigation is warranted to determine the relationship between p38 MAPK signaling and sPLA2 expression and activation.
Regulation of glutamate-induced inflammation
Following SCI, the glutamate level rises in the extracellular space, resulting in significant tissue damage (Liu et al., 1991, 1999; McAdoo et al., 1999). NO and PLA2, known targets of MH treatment, have been shown to inhibit astrocytic glutamate reuptake (Volterra et al., 1994) and contribute to elevated glutamate levels. Glutamate has been shown to induce microglial activation and proliferation in a p38 MAPK-dependent manner, resulting in IL-1β and NO release and neuronal apoptosis, while MH treatment was shown to abrogate this effect by inhibiting microglial p38 MAPK phosphorylation (Tikka et al., 2001). These findings highlight the complex interconnections between excitotoxicity, inflammatory signaling, and oxidative stress following SCI that could potentially serve as conserved mechanisms of MH activity in multiple contexts.
Mechanisms of Anti-Oxidative Activity
Oxidative damage occurs when cells are exposed to free radicals including ROS and RNS. The reactive species destabilize cell membranes, damage organelles, proteins and nucleic acids, and trigger apoptotic or necrotic pathways resulting in cell death (Ryter et al., 2007). The widespread oxidative damage induced by the highly reactive ROS/RNS may be central in the etiology of cellular death and functional loss after SCI (Oyinbo, 2011). Following SCI, disruption of the blood-spinal cord barrier results in hemorrhage and ischemia (Mautes et al., 2000). Under ischemic conditions, many cells die due to energetic failure, buildup of acidic anaerobic metabolites, loss of ionic homeostasis and mitochondrial dysfunction, while surviving cells produce excessive reactive species upon re-oxygenation of the tissue (Kalogeris et al., 2012). Furthermore, as blood-derived leukocytes enter the spinal cord tissueviadamaged blood vessels, they produce ROS/RNS (Trivedi et al., 2006), while blood-derived iron catalyzes lipid peroxidation reactions, yielding additional free radicals (Hall, 2011). In addition, glutamate activation of NMDA receptors increases intracellular Ca2+level, which activates neuronal NO synthase (nNOS)viacalmodulin (Conti et al., 2007). Later, microglia and astrocytes within the spinal cord become activated and upregulate iNOS and NADPH oxidase that are responsible for prolonged free radical production and tissue damage (Xu et al., 2005; Conti et al., 2007; Cooney et al., 2014). Early upregulation of nNOS after SCI has been suggested to be detrimental by increasing the oxidative stress in the injured spinal cord (Conti et al., 2007). Studies have shown that inhibition of nNOS may promote neuroprotection after SCI (Sharma et al., 2005; Sharma, 2010). In addition, inhibition of nNOS expression in motoneurons has been shown to increase their survival after spinal root avulsion (Wu et al., 2003; Sim et al., 2015). Additional sources of reactive species following SCI include glutamate-induced mitochondrial dysfunction, increased oxygen consumption and superoxide production by phago-cytic cells, as well as release of cytosolic oxidases, lysosomes, peroxisomes, and other cell constituents from necrotic cells (Jia et al., 2012). As a result, ROS/RNS production increases significantly, contributing to inflammation and resulting in widespread damage to both the cells producing free radicals and surrounding tissue (Visavadiya et al., 2016). MH can reduce reactive species production from activated microglia and macrophagesviapreviously discussed anti-inflammatory mechanisms. In addition, MH has been shown to inhibit thrombin and Zinc-induced activation of NADPH oxidase from reactive microglia, as evidenced by reduced translocation of NADPH oxidase subunit p67phox, an indicator of active NADPH oxidase assembly required for superoxide radical production (Kumar et al., 2015). However, the mechanism of this inhibition was not reported. Additionally, MH can attenuate glutamate-induced free radical productionviaits neuroprotective effects (Garcia-Martinez et al., 2010), which will be discussed in the neuroprotection section. Moreover, MH can directly scavenge free radicals, which will be discussed in this section. The anti-oxidative activity is an important mechanism by which MH can mitigate secondary injury progression.
Direct free radical scavenging activity
In addition to inhibiting free radical productionviaits anti-inflammatory and neuroprotective effects, MH can act as a phenolic antioxidant to directly eliminate free radicals in the post-injury microenvironment. It has been shown to exhibit powerful free-radical scavenging activity (Kraus et al., 2005) due to its phenol ring structure (Figure 2, red box). Free radicals can remove the hydrogen atom from the phenolic hydroxyl group in MH molecules, resulting in a phenol-derived free radical that is far less reactive due to resonance stabilization and steric hindrance around the phenol group (Kraus et al., 2005). MH was found to directly scavenge free radicals including DPPH (2,2-diphenyl-1-picrylhydrazyl) and ABTS [2,2-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammoniumsalt], reduce deoxyribose degradation by Fe2+/ascorbic acid/H2O2, and inhibit iron-induced lipid peroxidation (Kraus et al., 2005). Furthermore, studies have shown that MH can significantly inhibit lipid peroxidation after SCI (Sonmez et al., 2013; Aras et al., 2015). Treatment with MH resulted in decreased levels of malondialdehyde (MDA) (Sonmez et al., 2013; Aras et al., 2015), a byproduct of lipid peroxidation, and increased levels of glutathione (GSH), an endogenous antioxidant that neutralizes reactive species (Sonmez et al., 2013). MH treatment after SCI also led to increased activity of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px), enzymes responsible for neutralizing free radicals (Aras et al., 2015). GSH-Px catalyzes the neutralizing reaction between reactive species and GSH, yielding a glutathione disulfide species. Thus, decreases in GSH suggest increases in reactive species concentration and subsequent GSH consumption. Similarly, increases in GSH-Px and SOD activity could suggest the presence of free enzymes not actively catalyzing neutralization reactions, because of reduced levels of reactive species. In addition to SCI, MH has been shown to reduce glutamate-induced oxidative stress in neuronal cultures (Kraus et al., 2005), attenuate oxidative stress in a model of ischemia (Morimoto et al., 2005), and protect against oxidative damage in the brains of animals challenged with chronic mild stress (Réus et al., 2015). Taken together, these data illustrate powerful anti-oxidative mechanisms by which MH can reduce secondary injury after SCI, likely through both reduced free radical production and direct free-radical scavenging. Further investigation is warranted to determine the relative contributions of each aspect of anti-oxidative activity.
Mechanisms of Neuroprotection
As we have discussed, MH can reduce the toxicity of the post-SCI environment by inhibiting the production of neurotoxic molecules through modulating inflammation and scavenging free radicals. In addition, MH can directly protect neurons and glial cells from the neurotoxic environment after SCI (Elewa et al., 2006; Plane et al., 2010). In this section, we discuss the potential mechanisms of its direct neuroprotective effects.
Protection against glutamate excitotoxicity
Glutamate excitotoxicity is one of the major secondary injury mechanisms. A number of factors contribute to elevated glutamate levels in the injured spinal cord, including enhanced presynaptic glutamate efflux from injured neurons, reduction of glutamate uptake by astrocytes, and reverse Na+/glutamate transporter activity due to excess Na+ion buildup downstream of ATP synthase failure (Nishizawa, 2001; Park, 2004). Excessive glutamate activates ionotropic glutamate receptors, triggering the opening of associated ion channels and subsequent Ca2+influx (Park, 2004). Activation of glutamate receptor also results in suppression of PI3K/ Akt activation and subsequent p38 MAPK phosphorylation in cerebellar granule neurons (Pi et al., 2004), and increases downstream neuronal expression and activation of p38 MAPK pathway-associated pro-inflammatory and ROS-generating genes implicated in excitotoxic injury progression, including cPLA2, NO synthase, and NADPH oxidase (Dugan et al., 1995; Mark et al., 2001; Shen et al., 2007; Demaurex and Scorrano, 2009). Excessive Ca2+influx also results in mitochondrial Ca2+overload and has direct consequences on mitochondrial function, including uncoupling of electron transfer from ATP synthesis and resultant energy failure (Schinder et al., 1995; Kanki et al., 2004). When mitochondria become overloaded with Ca2+ions, the mitochondrial permeability transition pore (mPTP) opens (Ankarcrona et al., 1995), which subsequently triggers massive depolarization of mitochondrial membranes. This results in ATP deficiency and cytosolic release of mitochondrial contents including CytC (Norenberg and Rao, 2007), a crucial mediator of both apoptotic and necrotic cell death pathways (Bobba et al., 2002; Rasola and Bernardi, 2011). In addition, increased intracellular Ca2+activates calpains, a family of Ca2+-dependent cysteine proteases following SCI (Ray et al., 1999, 2003,2011). Overactivation of calpains degrades cytoskeletal and membrane proteins, resulting in both necrotic and apoptotic death after neuronal injury (Vosler et al., 2008; Ray et al., 2011). Additionally, calpains have been suggested to act synergistically with caspase-3 activation to promote apoptosis (Ray et al., 2001; Wingrave et al., 2003). Calpains can also promote cell death through an alternative caspase-independent mechanism mediated by mitochondrial release of CytC and apoptosis-inducing factor (Lankiewicz et al., 1999; Volbracht et al., 2005).

Figure 1 Inflammatory pathways involved in the anti-inflammatory action of MH.
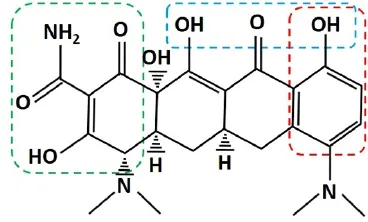
Figure 2 Chemical structure of minocycline hydrochloride (MH).
MH has been shown to protect cultured spinal cord- and brain-derived neurons from excitotoxic insult (Tikka et al., 2001; Tikka and Koistinaho, 2001; Gonzalez et al., 2007; Garcia-Martinez et al., 2010). Multiple mechanisms of protection have been suggested and a detailed illustration of neuroprotective mechanisms of MH against excitotoxicity is presented in Figure 3. MH has been shown to inhibit Ca2+influx through NMDA-responsive glutamate receptors (Garcia-Martinez et al., 2010). MH can chelate divalent and trivalent metal ions such as Ca2+, Mg2+, Zn2+, Fe2+and Fe3+(Figure 2) (Lambs et al., 1984; Grenier et al., 2000; Bauer et al., 2004; Chen-Roetling et al., 2009; Huang et al., 2012; Venkat et al., 2013; Wang et al., 2017). This property can theoretically reduce extracellular Ca2+concentration and thereby Ca2+influx. However, when cerebellar granule neurons were challenged with NMDA, MH was found to significantly reduce Ca2+influx; but when cells were challenged with high K+medium triggering voltage-gated Ca2+channels, no effect from MH treatment was observed (Garcia-Martinez et al., 2010). Because MH chelation occurred in both contexts, this illustrates that metal ion chelation is insufficient to reduce Ca2+influx. Instead, MH is likely interacting specifically with NMDA receptors. MH has been described as an NMDA receptor modulator (Chaves et al., 2009), and shown to modulate NMDA receptor signaling in hippocampal neurons (Gonzalez et al., 2007), but the exact nature of MH interaction with NMDA receptors remains poorly understood. Further investigation is warranted to elucidate a clear mechanism of action.

Figure 3 MH inhibits glutamate excitotoxicity in neurons.
In addition to inhibiting Ca2+influx through the cell membrane, MH was found to reduce mitochondrial Ca2+uptake by slightly depolarizing mitochondria, reducing the electrochemical gradient required for mitochondrial Ca2+uptake (Garcia-Martinez et al., 2010). The authors attributed this function to selective partial inhibition of electron transport chain complexes I and IV and modulation of the voltage-dependent anion channel (VDAC) (Garcia-Martinez et al., 2010). In addition, MH can inhibit the mitochondrial permeability transition. It is likely a result of its inhibition of mitochondrial Ca2+overload and oxidative stress, prerequisites for mPTP opening (Norenberg and Rao, 2007; Webster, 2012). MH has also been shown to inhibit mitochondrial Ca2+uptake in liver cells following ischemic insult (Theruvath et al., 2008; Schwartz et al., 2013), indicating a conserved mechanism of action across multiple cell types.
MH has been shown to inhibit the opening of the mPTP (Gieseler et al., 2009) and mitochondrial CytC releasein vivo(Zhu et al., 2002; Teng et al., 2004). Although mitochondrial release of CytC is often associated with increased mitochondrial permeability, it can also occur through other mechanisms (Bossy-Wetzel et al., 1998). For example, activation of upstream proapoptotic factors also causes CytC release (Stirling et al., 2005). When CytC is released from mitochondria, it initiates pro-apoptotic caspase signaling cascades, resulting in cell death (Cai et al., 1998). MH has been shown to inhibit CytC release and improve functional outcomes in rodent models of SCI and amyotrophic lateral sclerosis (Zhu et al., 2002; Teng et al., 2004). In addition, MH has been shown to attenuate increases in caspase-1 and 3 expressionin vivo(Chen et al., 2000; Festoff et al., 2006). Although the mechanism is not well understood, reduction in CytC release is likely a result of previously described MH activity in mitochondria, while inhibition of caspase expression could be a downstream effect of reduced CytC release.
MH can also inhibit NMDA-induced ROS production in cultured neurons (Garcia-Martinez et al., 2010). In addition to damaging proteins, lipids, and nucleic acids, ROS can directly damage mitochondria and induce mPTP opening as well (Dong et al., 2009). Reduced ROS production is likely a result of inhibition of p38 MAPK pathways, since upregulation of ROS-producing enzymes was previously shown to be dependent on p38 MAPK activation in other cell types. MH has been shown to inhibit p38 MAPK activation in neurons challenged with glutamate (Pi et al., 2004).
MH may also protect against excitotoxicity through modulation of PLA2 expression and activity, as cPLA2 has been implicated in excitotoxic progression in cultured neurons (Shen et al., 2007; Zhao et al., 2011b). In addition, activated cPLA2 was found in neurons following SCI, and implicated in injury progression (Liu et al., 2014). In neuronal cultures, cPLA2 activation has been shown to be regulated by p38 MAPK (Kriem et al., 2005), a known target of MH treatment in neurons (Pi et al., 2004). It follows that MH treatment likely reduces cPLA2 activation in neurons through inhibition of p38 MAPK pathway.
Enzymatic inhibition of PARP-1
Cell death following SCI is also associated with overactivation of poly(ADP-ribose) polymerase-1 (PARP-1) (Genovese and Cuzzocrea, 2008). PARP-1 is a nuclear enzyme implicated in DNA repair in healthy tissue, but can play a pathogenic role in response to excitotoxic insult and oxidative stress (Mandir et al., 2000; Ying et al., 2001; Du et al., 2003). PARP-1 activation is triggered by DNA damage (D’Amours et al., 1999). When the damage is mild, PARP-1 facilitates cell survival. However, severe DNA damage can induce excessive activation of PARP-1 (Wu et al., 2015), which depletes cytosolic nicotinamide adenine dinucleotide (NAD+), resulting in ATP-deficient energy failure and massive mitochondrial depolarization (Baxter et al., 2014). PARP-1 also induces mitochondrial release of apoptosis inducing factor (AIF), which translocates to the nucleus, leading to cell death(Wang et al., 2009). In addition, PARP-1 activation has been implicated in astrocyte activation—treatment of bacteria-stimulated astrocytes with a synthetic PARP-1 inhibitor resulted in reduced expression of IL-1β, TNFα, NO, and MCP-1 (Phulwani and Kielian, 2008). MH has been shown to effectively inhibit PARP-1 enzymatic activity under cell-free conditions in a dose-dependent manner, and to protect neurons against PARP-1 mediated death (Alano et al., 2006). The authors noted that the carboxyamide functional groups attached to aromatic rings are conserved among multiple known PARP inhibitors, and are present in MH’s chemical structure, indicating a possible structural basis for its activity to inhibit PARP-1 (Figure 2). Additionally, MH treatment was found to reduce abnormal PARP-1 activation in a rodent diabetic retinopathy model (Wu et al., 2015). PARP-1 inhibition is one of the major neuroprotective targets in therapeutic applications. Further studies are warranted to elucidate the role of MH-mediated PARP-1 inhibition following SCI and other neurological deficits.
Inhibition of MMPs
MH has been shown to inhibit MMPs, a class of metal-ion dependent enzymes capable of digesting extracellular matrix proteins. Multiple MMPs are rapidly upregulated following SCI, and are involved in both injury and recovery processes (Zhang et al., 2011). MMP-9, one of the key MMPs involved in secondary injury progression, plays an important role in breakdown of blood-spinal cord barrier, resulting in edema and invasion of peripheral immune cells and blood-derived components. MMP-related infiltration of blood-derived factors and immune cells results in increased inflammation, oxidative stress and apoptosis after SCI (Noble et al., 2002; Zhang et al., 2011). In rat models of SCI, infiltrating leukocytes were found to be the predominant source of MMP-9 activity (de Castro et al., 2000), while treatment with an MMP-2/MMP-9 inhibitor was shown to significantly reduce barrier disruption and apoptotic cell death (Yu et al., 2008). MH has been shown to inhibit both MMP-2 and MMP-9 activitiesin vitro, with a more potent effect on MMP-9 (Paemen et al., 1996; Machado et al., 2006; Modheji et al., 2016). MH inhibition of MMP-9 has also been illustrated in animal models of stroke, cardiomyopathy, cerebral ischemia, and fragile-X syndrome (Koistinaho et al., 2005; Machado et al., 2006; Bilousova et al., 2009; Matsumoto et al., 2009). MH has been found to inhibit MMP-9 activity under cell free conditions (Paemen et al., 1996), indicating that it can directly inhibit the enzymatic activities of MMPs. It has been suggested that MH could inhibit MMP activity by interacting with Zn2+ions that are critical for enzymatic activity (Golub et al., 1991; Griffin et al., 2010; Modheji et al., 2016). In a study involving multiple tetracycline derivatives, a positive correlation has been reported between tetracycline derivative affinity for Zn2+ions and MMP inhibition, whereas addition of excess Zn2+ions was shown to partially reverse inhibition of MMPs (Ryan et al., 2001). Thus, MH likely inhibits MMP activityviadirect inhibition of the enzyme, by interacting with metal ion moieties.
Protection against blood-derived iron toxicity
Following SCI, blood-derived factors and cells infiltrate the spinal cord tissueviathe disrupted blood-spinal cord barrier. Iron, a key blood component, has been shown to exert neurotoxic effects by catalyzing the formation of free radicalsviathe Fenton reaction (Winterbourn, 1995), resulting in subsequent lipid peroxidation and nucleic acid damage (Salvador et al., 2010; Núñez et al., 2012). In addition to mitigating the extent of disruption to the blood-brain barrier, MH has been shown to reduce iron neurotoxicity bothin vitroandin vivo(Kraus et al., 2005; Chen-Roetling et al., 2009; Zhao et al., 2011a). MH can reduce lipid peroxidation initiated by both Fe2+and Fe3+which can be found in the blood (Hall, 2011; Ebrahimi et al., 2013),viaits anti-oxidative activity (Kraus et al., 2005). In this study, MH reduced lipid peroxidation through a chelation-independent, free-radical scavenging mechanism (Kraus et al., 2005). MH has also been shown to inhibit iron neurotoxicity in cultured cortical neurons (Chen-Roetling et al., 2009). In this study, the protective effect was attributed to iron chelation, increased ferritin expression, and decreased iron-catalyzed lipid peroxidation (Chen-Roetling et al., 2009). Ferritin produced in response to iron overload can attenuate toxic iron levels, resulting in a protective reduction of iron concentration (Salvador, 2010).
Systemic administration of MH in a stroke model was shown to attenuate total serum iron levels, mitigate bloodbrain barrier disruption, reduce iron-overload in the brain, and attenuate neuronal death (Zhao et al., 2011a). In this study, MH actually reduced ferritin expression in the brain, likely a result of decreased serum iron levels, BBB disruption and iron infiltration into the CNS (Zhao et al., 2011a). Because MH can chelate both Fe2+and Fe3+(Bauer et al., 2004; Chen-Roetling et al., 2009; Huang et al., 2012), it is possible that MH can inhibit iron-mediated toxicity partially through iron chelationin vivo. Primary mechanisms of direct neuroprotective action against iron toxicity, however, are likely scavenging of iron-initiated free radicals and increased ferritin expression. While in the absence of irons MH did not significantly alter ferritin production in cortical neurons, exposure to irons induced a 10-fold increase in ferritin expression, and MH and iron co-treatment induced a 17-fold increase in ferritin expression (Chen-Roetling et al., 2009). This illustrates a potential antioxidant-independent mechanism by which MH alters the neuronal response to neurotoxic iron. Further investigation is warranted to determine the mechanism by which MH can induce upregulation of neuronal ferritin expression following iron insult.
Conclusions
Minocycline exhibits potent anti-inflammatory, anti-oxidative, and neuroprotective activities after SCI. Its anti-inflammatory and neuroprotective activities are partially achieved through conserved mechanisms such as modulation of p38 MAPK and PI3K/Akt signaling pathways and inhibition of MMPs. In addition, MH directly inhibits sPLA2, which is involved in conversion of AA into prostaglandins and leukotrienes. Both lipids are potent mediators of inflammationand secondary injury after SCI. The neuroprotective effects of MH are achieved through multiple mechanisms. Besides targeting p38 MAPK and PI3K/Akt signaling as well as MMPs, MH can also protect against glutamate exitotoxicity by diminishing Ca2+influx through the NMDA receptor into neurons and reducing mitochondrial Ca2+uptake. In addition, MH can exert neuroprotective effects by directly inhibiting the activities of neurotoxic molecules. For example, MH can inhibit PARP-1 enzymatic activityviathe carboxyamide functional groups attached to its aromatic rings. Furthermore, MH is a potent antioxidant. It can directly scavenge free radicals through the phenolic hydroxyl group. Because it can target many secondary injury mechanisms, MH holds great promise for the development of an effective therapy for SCI. Further research is warranted to determine the therapeutic window, as well as optimal dose, duration, and route of MH administration to achieve maximal benefit.
Author contributions:RBS and YZ wrote and finalized the paper.
Conflicts of interest:The authors declare no financial conflicts of interest.
Open peer reviewer:Jafri Malin Abdullah.
Additional file:Open peer review report 1.
Alano CC, Kauppinen TM, Valls AV, Swanson RA (2006) Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci U S A 103:9685-9690.
Altavilla D, Squadrito F, Bitto A, Polito F, Burnett BP, Di Stefano V, Minutoli L (2009) Flavocoxid, a dual inhibitor of cyclooxygenase and 5-lipoxygenase, blunts pro-inflammatory phenotype activation in endotoxin-stimulated macrophages. Br J Pharmacol 157:1410-1418.
Amin A, Attur M, Thakker G, Patel P, Vyas P, Patel R, Patel I, Abramson S (1996) A novel mechanism of action of tetracyclines: Effects on nitric oxide synthases. Proc Natl Acad Sci U S A 93:14014-14019.
Ankarcrona M, Dypkukt J, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton S, Nicotera P (1995) Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15:961-973.
Aras M, Altas M, Motor S, Dokuyucu R, Yilmaz A, Ozgiray E, Seraslan Y, Yilmaz N (2015) Protective effects of minocycline on experimental spinal cord injury in rats. Injury 46:1471-1474.
Balsinde J, Winstead M, Dennis E (2002) Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett 531:2-6.
Bauer G, Berens C, Projan SJ, Hillen W (2004) Comparison of tetracycline and tigecycline binding to ribosomes mapped by dimethylsulphate and drug-directed Fe2+cleavage of 16S rRNA. J Antimicrob Chemother 53:592-599.
Baxter P, Chen Y, Xu Y, Swanson RA (2014) Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: a treatable cause of cell death in stroke. Transl Stroke Res 5:136-144.
Beattie MS, Harrington A, Lee R, Kim J, Boyce S, Longo F, Bresnahan J, Hempstead B, Ok Yoon S (2002) ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36:375-386.
Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM (2009) Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet 46:94-102.
Bobba A, Canu N, Atlante A, Petragallo V, Calissano P, Marra E (2002) Proteasome inhibitors prevent cytochrome c release during apopotosis but not in excitotoxic death of cerebellar granule neurons. FEBS Lett 515:8-12.
Boden S, Bertsche T, Ammon H, Safayhi H (2000) MEK-1/2 inhibition prevents 5-lipoxygenase translocation in N-formylpeptide-challenged human neutrophils. Int J Biochem Cell Biol 32:1069-1074.
Bossy-Wetzel E, Newmeyer D, Green D (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J 17:37-49.
Buczynski MW, Svensson CI, Dumlao DS, Fitzsimmons BL, Shim JH, Scherbart TJ, Jacobsen FE, Hua XY, Yaksh TL, Dennis EA (2010) Inflammatory hyperalgesia induces essential bioactive lipid production in the spinal cord. J Neurochem 114:981-993.
Byrnes KR, Garay J, Di Giovanni S, De Biase A, Knoblach SM, Hoffman EP, Movsesyan V, Faden AI (2006) Expression of two temporally distinct microglia-related gene clusters after spinal cord injury. Glia 53:420-433.
Cai J, Yang J, Jones D (1998) Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta 1366:139-149.
Cai Z, Yan Y, Chen R (2010) Minocycline reduces astrocytic reactivation and neuroinflammation in the hippocampus of a vascular cognitive impairment rat model. Neurosci Bull 26:28-36.
Ceccarelli S, Panera N, Mina M, Gnani D, De Stefanis C, Crudele A, Rychlicki C, Petrini S, Bruscalupi G, Agostinelli L, Stronati L, Cucchiara S, Musso G, Furlanello C, Svegliati-Baroni G, Nobili V, Alisi A (2015) LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 6:41434-41452.
Chaves C, Marque C, Trzesniak C, Machado de Sousa J, Zuardi A, Crippa J, Dursun S, Hallak J (2009) Glutamate-N-methyl-D-aspartate receptor modulation and minocycline for the treatment of patients with schizophrenia: an update. Braz J Med Biol Res 42:1002-1014.
Chen B, Chen Y, Lin W (1999) Involvement of p38 mitogen-activated protein kinase in lipopolysaccharide-induced iNOS and COX-2 expression in J774 macrophages. Immunology 97:124-129.
Chen M, Ona V, Li M, Ferrante R, Fink K, Zhu S, Bian J, jGui L, Farrell L, Hersch S, Hobbs W, Vonsattel J, Cha J, Friedlander RM (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6:797-801.
Chen-Roetling J, Chen L, Regan RF (2009) Minocycline attenuates iron neurotoxicity in cortical cell cultures. Biochem Biophys Res Commun 386:322-326.
Chin TY, Kiat SS, Faizul HG, Wu W, Abdullah JM (2017) The effects of minocycline on spinal root avulsion injury in rat model. Malays J Med Sci 24:31-39.
Choi SH, Lee DY, Chung ES, Hong YB, Kim SU, Jin BK (2005) Inhibition of thrombin-induced microglial activation and NADPH oxidase by minocycline protects dopaminergic neurons in the substantia nigra in vivo. J Neurochem 95:1755-1765.
Chu LS, Fang SH, Zhou Y, Yu GL, Wang ML, Zhang WP, Wei EQ (2007) Minocycline inhibits 5-lipoxygenase activation and brain inflammation after focal cerebral ischemia in rats. Acta Pharmacol Sin 28:763-772.
Chu LS, Fang SH, Zhou Y, Yin YJ, Chen WY, Li JH, Sun J, Wang ML, Zhang WP, Wei EQ (2010) Minocycline inhibits 5-lipoxygenase expression and accelerates functional recovery in chronic phase of focal cerebral ischemia in rats. Life Sci 86:170-177.
Conti A, Miscusi M, Cardali S, Germanò A, Suzuki H, Cuzzocrea S, Tomasello F (2007) Nitric oxide in the injured spinal cord: synthases cross-talk, oxidative stress and inflammation. Brain Res Rev 54:205-218.
Cooney SJ, Zhao Y, Byrnes KR (2014) Characterization of the expression and inflammatory activity of NADPH oxidase after spinal cord injury. Free Radic Res 48:929-939.
Corsaro A, Thellung S, Chiovitti K, Villa V, Simi A, Raggi F, Paludi D, Russo C, Aceto A, Florio T (2009) Dual modulation of ERK1/2 and p38 MAP kinase activities induced by minocycline reverses the neurotoxic effects of the prion protein fragment 90-231. Neurotox Res 15:138-154.
Coulon L, Calzada C, Moulin P, Véricel E, Lagarde M (2003) Activation of p38 mitogen-activated protein kinase/cytosolic phospholipase A2 cascade in hydroperoxide-stressed platelets. Free Radic Biol Med 35:616-625.
D’Amours D, Desnoyers S, D’Silva I, Poirier G (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342:249-268.
Dalm D, Palm GJ, Aleksandrov A, Simonson T, Hinrichs W (2010) Nonantibiotic properties of tetracyclines: structural basis for inhibition of secretory phospholipase A2. J Mol Biol 398:83-96.
David S, López-Vales R, Wee Yong V (2012) Harmful and beneficial effects of inflammation after spinal cord injury: potential therapeutic implications. Handb Clin Neurol 109:485-502.
de Castro RJ, Burns C, McAdoo D, Romanic A (2000) Metalloproteinase increases in the injured rat spinal cord. Neuroreport 11:3551-3554.
Dean J, Brook M, Clark A, Saklatvaia J (1999) p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem 274:264-269.
Demaurex N, Scorrano L (2009) Reactive oxygen species are NOXious for neurons. Nat Neurosci 12:819-820.
Di Filippo M, de Iure A, Giampa C, Chiasserini D, Tozzi A, Orvietani PL, Ghiglieri V, Tantucci M, Durante V, Quiroga-Varela A, Mancini A, Costa C, Sarchielli P, Fusco FR, Calabresi P (2016) Persistent activation of microglia and NADPH oxidase drive hippocampal dysfunction in experimental multiple sclerosis. Sci Rep 6:20926.
Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30:379-387.
D’Orsi B, Bonner H, Tuffy LP, Dussmann H, Woods I, Courtney MJ, Ward MW, Prehn JH (2012) Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J Neurosci 32:1847-1858.
Dou F, Huang L, Yu P, Zhu H, Wang X, Zou J, Lu P, Xu XM (2009) Temporospatial expression and cellular localization of oligodendrocyte myelin glycoprotein (OMgp) after traumatic spinal cord injury in adult rats. J Neurotrauma 26:2299-2311.
Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, Clark RS (2003) Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem 278:18426-18433.
Dugan L, Sensi S, Canzoniero L, Handran S, Rothman S, Lin T, Goldberg M, Choi D (1995) Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci 15:6377-6388.
Dulin JN, Karoly ED, Wang Y, Strobel HW, Grill RJ (2013) Licofelone modulates neuroinflammation and attenuates mechanical hypersensitivity in the chronic phase of spinal cord injury. J Neurosci 33:652-664.
Ebrahimi K, Bill E, Hagedoorn P, Hagen W (2013) The catalytic center of ferritin regulates iron storage via Fe(II)-Fe(III) displacement. Nat Chem Biol 8:941-948.
Elewa HF, Hilali H, Hess DC, Machado LS, Fagan SC (2006) Minocycline for acute neuroprotection. Pharmacotherapy 26:515-521.
Fehlings MG, Nguyen DH (2010) Immunoglobulin G: a potential treatment to attenuate neuroinflammation following spinal cord injury. J Clin Immunol 30 Suppl 1:S109-112.
Festoff BW, Ameenuddin S, Arnold PM, Wong A, Santacruz KS, Citron BA (2006) Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem 97:1314-1326.
Filbin MT (2003) Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci 4:703-713.
Fitch M, Doller C, Combs C, Landreth G, Silver J (1999) Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 19:8182-8198.
Garcia-Martinez EM, Sanz-Blasco S, Karachitos A, Bandez MJ, Fernandez-Gomez FJ, Perez-Alvarez S, de Mera RM, Jordan MJ, Aguirre N, Galindo MF, Villalobos C, Navarro A, Kmita H, Jordan J (2010) Mitochondria and calcium flux as targets of neuroprotection caused by minocycline in cerebellar granule cells. Biochem Pharmacol 79:239-250.
Genovese T, Cuzzocrea S (2008) Role of free radicals and poly(ADP-ribose)polymerase-1 in the development of spinal cord injury: new potential therapeutic targets. Curr Med Chem 15:477-487.
Genovese T, Mazzon E, Rossi A, Di Paola R, Cannavo G, Muia C, Crisafulli C, Bramanti P, Sautebin L, Cuzzocrea S (2005) Involvement of 5-lipoxygenase in spinal cord injury. J Neuroimmunol 166:55-64.
Gensel JC, Zhang B (2015) Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res 1619:1-11.
Ghazali FH, Wu W, Abdullah JM (2016) Histological analysis of motoneuron survival and microglia inhibition after nerve root avulsion treated with nerve graft implantation and minocycline: an experimental study. Sains Malays 45:1641-1648.
Gieseler A, Schultze AT, Kupsch K, Haroon MF, Wolf G, Siemen D, Kreutzmann P (2009) Inhibitory modulation of the mitochondrial permeability transition by minocycline. Biochem Pharmacol 77:888-896.
Golub L, Ramamurthy N, McNamare T, Greenwalk R, Rifkin B (1991) Tetracyclines inhibit connective tissue breakdown: new therapeutic implications for an old family of drugs. Crit Rev Oral Biol Med 2:297-321.
Gonzalez JC, Egea J, Del Carmen Godino M, Fernandez-Gomez FJ, Sanchez-Prieto J, Gandia L, Garcia AG, Jordan J, Hernandez-Guijo JM (2007) Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca(2+) signalling in hippocampal neurons. Eur J Neurosci 26:2481-2495.
Gonzalez R, Glaser J, Liu MT, Lane TE, Keirstead HS (2003) Reducing inflammation decreases secondary degeneration and functional deficit after spinal cord injury. Exp Neurol 184:456-463.
Grenier D, Huot M, Mayrand D (2000) Iron-chelating activity of tetracyclines and its impact on the susceptibility of actinobacillus actinomycetemcomitans to these antibiotics. Antimicrob Agents Chemother 44:763-766.
Griffin M, Fricovsky E, Ceballos G, Villarreal F (2010) Tetracyclines: a pleitropic family of compounds with promising therapeutic properties. Review of the literature. Am J Physiol Cell Physiol 299:C539-548.
Guha M, Mackman N (2002) The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem 277:32124-32132.
Hacke K, Rincon-Orozco B, Buchwalter G, Siehler S, Wasylyk B, Wiesmüller L, Rösl F (2010) Regulation of MCP-1 chemokine transcription by p53. Mol Cancer 9:82.
Hahn J, Kaushik D, Mishra M, Wang J, Silva C, Yong V (2016) Impact of minocycline on extracellular matrix metalloproteinase inducer, a factor implicated in multiple sclerosis immunopathogenesis. J Immunol 197:3850-3860.
Hall ED (2011) Antioxidant therapies for acute spinal cord injury. Neurotherapeutics 8:152-167.
Hempstead BL (2009) Commentary: Regulating proNGF action: multiple targets for therapeutic intervention. Neurotox Res 16:255-260.
Hernández M, Bayón Y, Sánchez C, Nieto M (1999) Signaling mechanisms involved in the activation of arachidonic acid metabolism in human astrocytoma cells by tumor necrosis factor-alpha: phosphorylation of cytosolic phospholipase A2 and transactivation of cyclooxygenase-2. J Neurochem 73:1641-1649.
Hirose N, Maekawa T, Shinigawa T, Ishli S (2009) ATF-2 regulates lipopolysaccharide-induced transciption in macrophage cells. Biochem Biophys Res Commun 385:72-77.
Huang C, Lee Y, Yang Y, Kuo T, Huang N (2012) Minocycline prevents paraquat-induced cell death through attenuating endoplasmic reticulum stress and mitochondrial dysfunction. Toxicol Lett 209:203-210.
Jia Z, Zhu H, Li J, Wang X, Misra H, Li Y (2012) Oxidative stress in spinal cord injury and antioxidant-based intervention. Spinal Cord 50:264-274.
Kalogeris T, Baines CP, Krenz M, Korthuis RJ (2012) Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298:229-317.
Kanki R, Nakamizo T, Yamashita H, Kihara T, Sawada H, Uemura K, Kawamata J, Shibasaki H, Akaike A, Shimohama S (2004) Effects of mitochondrial dysfunction on glutamate receptor-mediated neurotoxicity in cultured rat spinal motor neurons. Brain Res 1015:73-81.
Kielian T, Esen N, Liu S, Phulwani NK, Syed MM, Phillips N, Nishina K, Cheung AL, Schwartzman JD, Ruhe JJ (2007) Minocycline modulates neuroinflammation independently of its antimicrobial activity in staphylococcus aureus-induced brain abscess. Am J Pathol 171:1199-1214.
Koistinaho M, Malm TM, Kettunen MI, Goldsteins G, Starckx S, Kauppinen RA, Opdenakker G, Koistinaho J (2005) Minocycline protects against permanent cerebral ischemia in wild type but not in matrix metalloprotease-9-deficient mice. J Cereb Blood Flow Metab 25:460-467.
Korhonen R, Lahti A, Kankaanranta H, Moilanen E (2005) Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy 4:471-479.
Krady J, Basu A, Allen C, Xu Y, LaNoue K, Gardner T, Levison S (2005) Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 54:1559-1565.
Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW (2005) Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem 94:819-827.
Kriem B, Sponne I, Fifre A, Malaplate-Armand C, Lozac’h-Pillot K, Koziel V, Yen-Potin F, Bihain B, Oster T, Olivier J, Pillot T (2005) Cytosolic phospholipase A2 mediates neuronal apoptosis induced by soluble oligomers of the amyloid-beta peptide. FASEB J 19:85-87.
Kumar V, Singh BK, Chauhan AK, Singh D, Patel DK, Singh C (2016) Minocycline rescues from zinc-induced nigrostriatal dopaminergic neurodegeneration: biochemical and molecular interventions. Mol Neurobiol 53:2761-2777.
Lambs L, Brion M, Berthon G (1984) Metal ion-tetracycline interactions in biological fluids. Part 3. Formation of mixed-metal ternary complexes of tetracycline, oxytetracycline, doxycycline and minocycline with calcium and magnesium, and their involvement in the bioavailability of these antibiotics in blood plasma. Agents Actions 14:743-750.
Lammertse DP (2013) Clinical trials in spinal cord injury: lessons learned on the path to translation. The 2011 International Spinal Cord Society Sir Ludwig Guttmann Lecture. Spinal Cord 51:2-9.
Lankiewicz S, Luetjens CM, Bui NT, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH (1999) Activation of calpain i converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem 275:17064-17071.
Lee S, Yune T, Kim S, Park D, Lee Y, Kim Y, Oh Y, Markelonis G, Oh T (2003a) Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma 20:1017-1027.
Lee SM, Yune TY, Kim SJ, Park DW, Lee YK, Kim YC, Oh YJ, Markelonis GJ, Oh TH (2003b) Minocycline reuces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma 20:1017-1027.
Liu D, Thangnipon W, McAdoo D (1991) Excitatory amino acids rise to toxic levels upon impact injury to the rat spinal cord. Brain Res 547:344-348.
Liu D, Xu G, Pan E, McAdoo D (1999) Neurotoxicity of glutamate at the concentration released upon spinal cord injury. Neuroscience 93:1383-1389.
Liu NK, Xu XM (2010) Phospholipase A2 and its molecular mechanism after spinal cord injury. Mol Neurobiol 41:197-205.
Liu NK, Deng LX, Zhang YP, Lu QB, Wang XF, Hu JG, Oakes E, Bonventre JV, Shields CB, Xu XM (2014) Cytosolic phospholipase A2 protein as a novel therapeutic target for spinal cord injury. Ann Neurol 75:644-658.
Ma L, Nagai J, Ueda H (2010) Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. J Neurochem 115:643-653.
Machado LS, Kozak A, Ergul A, Hess DC, Borlongan CV, Fagan SC (2006) Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci 7:56.
Mandir A, Poitras M, Berliner A, Herring W, Guastella D, Feldman A, Poirier G, Wang Z, Dawson T, Dawson V (2000) NMDA but not non-NMDA excitotoxicity is mediated by poly(ADP-ribose) polymerase. J Neurosci 20:8005-8011.
Mark L, Prost R, Ulmer J, Smith M, Daniels D, Strottmann J, Brown W, Hacein-Bey L (2001) Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging. AJNR AM J Neuroradiol 22:1813-1824.
Matsumoto Y, Park IK, Kohyama K (2009) Matrix metalloproteinase (MMP)-9, but not MMP-2, is involved in the development and progression of C protein-induced myocarditis and subsequent dilated cardiomyopathy. J Immunol 183:4773-4781.
Mautes A, Weinzierl M, Donovan F, Noble L (2000) Vascular events after spinal cord injury: contribution to secondary pathogenesis. Phys Ther 80:673-687.
McAdoo D, XU G, Robak G, Hughes M (1999) Changes in amino acid concentrations over time and space around an impact injury and their diffusion through the rat spinal cord. Exp Neurol 159:538-544.
McTigue DM, Tani M, Krivacic K, Chernosky A, Kelner G, Maciejewski D, Maki R, Ransohoff R, Stokes BT (1998) Selective chemokine mRNA accumulation in the rat spinal cord after contusion injury. J Neurosci Res 53:368-376.
Modheji M, Olapour S, Khodayar MJ, Jalili A, Yaghooti H (2016) Minocycline is more potent than tetracycline and doxycycline in inhibiting MMP-9 in vitro. Jundishapur J Nat Pharm Prod 11:e27377.
Morimoto N, Shimazawa M, Yamashima T, Nagai H, Hara H (2005) Minocycline inhibits oxidative stress and decreases in vitro and in vivo ischemic neuronal damage. Brain Res 1044:8-15.
Nagano S, Otsuka T, Niiro H, Yamaoka K, Arinobu Y, Ogami E, Akahoshi M, Inoue Y, Miyake K, Nakashima H, Niho Y, Harade M (2002) Molecular mechanisms of lipopolysaccharide-induced cyclooxygenase-2 expression in human neutrophils: involvement of the mitogen-activated protein kinase pathway and regulation by anti-inflammatory cytokines. Int Immunol 14:733-740.
Newton R, Cambridge L, Hart L, Stevens D, Lindsay M, Barnes P (2000) The MAP kinase inhibitors, PD098059, UO126 and SB203580, inhibit IL-1b-dependent PGE2 release via mechanistically distinct processes. Br J Pharmacol 130:1353-1361.
Nishizawa Y (2001) Glutamate release and neuronal damage in ischemia. Life Sci 69:369-381.
Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH (2008) Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood-brain barrier disruption after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab 28:1686-1696.
Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z (2002) Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci 22:7526-7535.
Norenberg MD, Rao KV (2007) The mitochondrial permeability transition in neurologic disease. Neurochem Int 50:983-997.
Núñez M, Urrutia P, Mena N, Aguirre P, Tapia V, Salazar J (2012) Iron toxicity in neurodegeneration. Biometals 25:761-776.
Nykjaer A, Lee R, Teng K, Jansen P, Madsen P, Neilsen M, Jacobsen C, Kliemannel M, Schwarz E, Willnow T, Hempstead B, Peterson C (2004) Sortilin is essential for proNGF-induced neuronal cell death. Nature 627:843-848.
Olson CM, Hedrick MN, Izadi H, Bates TC, Olivera ER, Anguita J (2007) p38 mitogen-activated protein kinase controls NF-kappaB transcriptional activation and tumor necrosis factor alpha production through RelA phosphorylation mediated by mitogen- and stress-activated protein kinase 1 in response to Borrelia burgdorferi antigens. Infect and Immun 75:270-277.
Oyinbo CA (2011) Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp 71:281-299.
Paemen L, Martens E, Norga K, Masure S, Roets E, Hoogmartens J, Opdenakker G (1996) The gelatinase inhibitory activity of tetracyclines and chemically modified tetracycline analogues as measured by a novel microtiter assay for inhibitors. Biochem Pharmacol 52:105-111.
Pang T, Wang J, Benicky J, Saavedra JM (2012) Minocycline ameliorates LPS-induced inflammation in human monocytes by novel mechanisms including LOX-1, Nur77 and LITAF inhibition. Biochim Biophys Acta 1820:503-510.
Park E, Velumian AA, Fehlings MG (2004) The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma 21:754-774.
Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427-434.
Phulwani NK, Kielian T (2008) Poly(ADP-ribose) polymerases (PARPs) 1-3 regulate astrocyte activation. J Neurochem 106:578-590.
Pi R, Li W, Lee NT, Chan HH, Pu Y, Chan LN, Sucher NJ, Chang DC, Li M, Han Y (2004) Minocycline prevents glutamate-induced apoptosis of cerebellar granule neurons by differential regulation of p38 and Akt pathways. J Neurochem 91:1219-1230.
Pineau I, Lacroix S (2007) Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol 500:267-285.
Plane JM, Shen Y, Pleasure DE, Deng W (2010) Prospects for minocycline neuroprotection. Arch Neurol 67:1442-1448.
Pruzanski W, Greenwald R, Street I, Laliberte F, tefanski E, Vadas P (1992) Inhibition of enzymatic activity of phospholipases A2 by minocycline and doxycycline. Biochem Pharmacol 44:1165-1170.
Rasola A, Bernardi P (2011) Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50:222-233.
Ray SK, Matzelle DD, Sribnick EA, Guyton MK, Wingrave JM, Banik NL (2003) Calpain inhibitor prevented apoptosis and maintained transcription of proteolipid protein and myelin basic protein genes in rat spinal cord injury. J Chem Neuroanat 26:119-124.
Ray SK, Matzelle DD, Wilford GG, Hogan EL, Banik NL (2001) Inhibition of calpain-mediated apoptosis by E-64 d-reduced immediate early gene (IEG) expression and reactive astrogliosis in the lesion and penumbra following spinal cord injury in rats. Brain Res 916:115-126.
Ray SK, Samantaray S, Smith JA, Matzelle DD, Das A, Banik NL (2011) Inhibition of cysteine proteases in acute and chronic spinal cord injury. Neurotherapeutics 8:180-186.
Ray SK, Shields DC, Saido TC, Matzelle DC, Wilford GG, Hogan EL, Banik NL (1999) Calpain activity and translational expression increased in spinal cord injury. Brain Res 816:375-380.
Resnick D, Graham S, Dixon C, Marion D (1998) Role of cyclooxygenase 2 in acute spinal cord injury. J Neurotrauma 15:1005-1013.
Réus G, Abeliara H, Maciel A, Dos Santos M, Carlesi A, Steckert A, Ferrira G, De Prá S, Streck E, Macêdo D, Queveo J (2015) Minocycline protects against oxidative damage and alters energy metabolism parameters in the brain of rats subjecte to chronic mild stress. Metab Brain Dis 30:545-553.
Rosenson R, Gelb M (2009) Secretory phospholipase A2: a multifaceted family of proatherogenic enzymes. Curr Cardiol Rep 11:445-451.
Rossignol S, Schwab M, Schwartz M, Fehlings MG (2007) Spinal cord injury: time to move? J Neurosci 27:11782-11792.
Ryan M, Usman A, Ramamurthy N, Golub L, Greenwald R (2001) Excessive matrix metalloproteinase activity in diabetes: inhibition by tetracycline analogues with zinc reactivity. Curr Med Chem 8:305-316.
Ryter S, Kim H, Hoetzel A, Park J, Nakahira K, Wang X, Choi A (2007) Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 9:49-89.
Salvador GA, Uranga RM, Giusto NM (2010) Iron and mechanisms of neurotoxicity. Int J Alzheimers Dis 2011:720658.
Sapadin AN, Fleischmajer R (2006) Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol 54:258-265.
Schinder A, Olson E, Spitzer N, Montal M (1995) Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci 16:6125-6133.
Schwartz J, Holmuhamedov E, Zhang X, Lovelace GL, Smith CD, Lemasters JJ (2013) Minocycline and doxycycline, but not other tetracycline-derived compounds, protect liver cells from chemical hypoxia and ischemia/reperfusion injury by inhibition of the mitochondrial calcium uniporter. Toxicol Appl Pharmacol 273:172-179.
Schweigreiter R, Bandtlow C (2006) Nogo in the injured spinal cord. J Neurotrauma 23:384-396.
Sharma H, Olsson Y, Nyberg F, Dey P (1993) Prostaglandins modulate alterations of microvascular permeability, blood flow, edema and serotonin levels following spinal cord injury: an experimental study in the rat. Neuroscience 57:443-449.
Sharma HS, Badgaiyan RD, Alm P, Mohanty S, Wiklund L (2005) Neuroprotective effects of nitric oxide synthase inhibitors in spinal cord injury-induced pathophysiology and motor functions: an experimental study in the rat. Ann N Y Acad Sci 1053:422-434.
Sharma HS (2010) A combination of tumor necrosis factor-alpha and neuronal nitric oxide synthase antibodies applied topically over the traumatized spinal cord enhances neuroprotection and functional recovery in the rat. Ann N Y Acad Sci 1199:175-185.
Sharma J, Al-Omran A, Parvathy S (2007) Role of nitric oxide in inflammatory diseases. Inflammopharmacology 15:252-259.
Shen Y, Kishimoto K, Linden DJ, Sapirstein A (2007) Cytosolic phospholipase A(2) alpha mediates electrophysiologic responses of hippocampal pyramidal neurons to neurotoxic NMDA treatment. Proc Natl Acad Sci U S A 104:6078-6083.
Shields DC, Schaehcer KE, Hogan EL, Banik NL (2000) Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J Neurosci Res 61:146-150.
Sim SK, Tan YC, Tee JH, Yusoff AA, Abdullah JM (2015) Expression of neuronal nitric oxide synthase and prevents mitochondrial dysfunction in spinal ventral horn in rats after C7 spinal root avulsion. Turk Neurosurg 25:617-624.
Slomiany B, Slomiany A (2013) Involvement of p38 MAPK-dependent activator protein (AP-1) activation in modulation of gastric mucosal inflammatory responses to Helicobacter pylori by ghrelin. Inflammopharmacology 21:67-78.
Song ZP, Xiong BR, Guan XH, Cao F, Manyande A, Zhou YQ, Zheng H, Tian YK (2016) Minocycline attenuates bone cancer pain in rats by inhibiting NF-kappaB in spinal astrocytes. Acta Pharmacol Sin 37:753-762.
Sonmez E, Kabatas S, Ozen O, Karabay G, Turkoglu S, Ogus E, Yilmaz C, Caner H, Altinors N (2013) Minocycline treatment inhibits lipid peroxidation, preserves spinal cord ultrastructure, and improves functional outcome after traumatic spinal cord injury in the rat. Spine 38:1253-1259.
Spencer NG, Schilling T, Miralles F, Eder C (2016) Mechanisms underlying interferon-gamma-induced priming of microglial reactive oxygen species production. PLoS One 11:e0162497.
Stammers AT, Liu J, Kwon BK (2012) Expression of inflammatory cytokines following acute spinal cord injury in a rodent model. J Neurosci Res 90:782-790.
Stirling DP, Koochesfahani KM, Steevers JD, Tetzlaff W (2005) Minocycline as a neuroprotective agent. Neuroscientist 11:308-322.
Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W (2004) Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 24:2182-2190.
Sung CS, Cherng CH, Wen ZH, Chang WK, Huang SY, Lin SL, Chan KH, Wong CS (2012) Minocycline and fluorocitrate suppress spinal nociceptive signaling in intrathecal IL-1beta-induced thermal hyperalgesic rats. Glia 60:2004-2017.
Switzer JA, Sikora A, Ergul A, Waller JL, Hess DC, Fagan SC (2012) Minocycline prevents IL-6 increase after acute ischemic stroke. Transl Stroke Res 3:363-368.
Takeda M, Kawaguchi M, Kumatoriya T, Horiuchi T, Watanabe K, Inoue S, Konishi N, Furuya H (2011) Effects of minocycline on hindlimb motor function and gray and white matter injury after spinal cord ischemia in rats. Spine 36:1919-1924.
Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM (2004) Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A 101:3071-3076.
Theruvath TP, Zhong Z, Pediaditakis P, Ramshesh VK, Currin RT, Tikunov A, Holmuhamedov E, Lemasters JJ (2008) Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate storage/reperfusion injury after rat liver transplantation through suppression of the mitochondrial permeability transition. Hepatology 47:236-246.
Thuret S, Moon LD, Gage FH (2006) Therapeutic interventions after spinal cord injury. Nat Rev Neurosci 7:628-643.
Tikka T, Fiebich B, Goldstein G, Keinänen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21:2580-2588.
Tikka TM, Koistinaho JE (2001) Minocycline provides neuroprotection against N-Methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol 166:7527-7533.
Titsworth W, Liu N, Xu X (2008) Role of secretory phospholipase A2 in CNS inflammation- implications in traumatic spinal cord injury. CNS Neurol Disord Drug Targets 7:254-269.
Trivedi JV, Olivas A, Noble-Haeusslein L (2006) Inflammation and spinal cord injury: infiltrating leukocytes as determinants of injury and repair processes. Clin Neurosci Res 6:283-292.
Varma AK, Das A, Wallace Gt, Barry J, Vertegel AA, Ray SK, Banik NL (2013) Spinal cord injury: a review of current therapy, future treatments, and basic science frontiers. Neurochem Res 38:895-905.
Venkat A, Chaitanya P, Ravinder P, Shilendra D (2013) Protective role of Minocycline through zinc chelation in LPS induced Alzheimer’s model. JSRP 2:1-7.
Visavadiya NP, Patel SP, VanRooyen JL, Sullivan PG, Rabchevsky AG (2016) Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol 8:59-67.
Volbracht C, Chua BT, Ng CP, Bahr BA, Hong W, Li P (2005) The critical role of calpain versus caspase activation in excitotoxic injury induced by nitric oxide. J Neurochem 93:1280-1292.
Volterra A, Trotti D, Racagni G (1994) Glutamate uptake is inhibited by arachidonic acid and oxygen radicals via two distinct and additive mechanism. Mol Pharmacol 46:986-992.
Vosler PS, Brennan CS, Chen J (2008) Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol 38:78-100.
Wang Y, Dawson VL, Dawson TM (2009) Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol 218:193-202.
Wang Z, Nong J, Shultz RB, Zhang Z, Kim T, Tom VJ, Ponnappan RK, Zhong Y (2017) Local delivery of minocycline from metal ion-assisted self-assembled complexes promotes neuroprotection and functional recovery after spinal cord injury. Biomaterials 112:62-71.
Wasserman J, Schlichter L (2007) Minocycline protects the blood-brain barrier and reduces edema following intracerebral hemorrhage in the rat. Exp Neurol 207:227-237.
Waterman W, Molski T, Huang C, Adams J, Sha’afi R (1996) Tumour necrosis factor-α-induced phosphorylation and activation of cytosolic phospholipase A2 are abrogated by an inhibitor of the p38 mitogen-activated protein kinase cascade in human neutrophils. Biochem J 319:17-20.
Webster KA (2012) Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiol 8:863-884.
Werz O, Klemm J, Samuelsson B, Radmark O (2000) 5-Lipoxygenase is phosphorylated by p38 kinase-dependent MAPKAP kinases. Proc Natl Acad Sci U S A 97:5261-5266.
Wingrave JM, Schaecher KE, Sribnick EA, WIlford GG, Ray SK, Hazen-Martin DJ, Hogan EL, Banik NL (2003) Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J Neurosci Res 73:95-104.
Winterbourn CC (1995) Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 82/83:969-974.
Wu W, Li L, Yick LW, Chai H, Xie Y, Yang Y, Prevette DM, Oppenheim RW (2003) GDNF and BDNF alter the expression of neuronal NOS, c-Jun, and p75 and prevent motoneuron death following spinal root avulsion in adult rats. J Neurotrauma 20:603-612.
Wu Y, Chen Y, Wu Q, Jia L, Du X (2015) Minocycline inhibits PARP1 expression and decreases apoptosis in diabetic retinopathy. Mol Med Rep 12:4887-4894.
Xu J, Hsu CY, Liu T, Hogan E, Perot PJ, Tai H (1990) Leukotriene B4 release and polymorphonuclear cell infiltration in spinal cord injury. J Neurochem 55:907-912.
Xu W, Chi L, Xu R, Ke Y, Luo C, Cai J, Qiu M, Gozal D, Liu R (2005) Increased production of reactive oxygen species contributes to motor neuron death in a compression mouse model of spinal cord injury. Spinal Cord 43:204-213.
Yadav UC, Ramana KV (2013) Regulation of NF-kappaB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid Med Cell Longev 2013:690545.
Yang Y, Kim SC, Yu T, Yi YS, Rhee MH, Sung GH, Yoo BC, Cho JY (2014) Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediators Inflamm 2014:352371.
Ying W, Sevigny MB, Chen Y, Swanson RA (2001) Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc Natl Acad Sci U S A 98:12227-12232.
Yiu G, He Z (2006) Glial inhibition of CNS axon regeneration. Nat Rev Neurosci 7:617-627.
Yu F, Kamada H, Niizuma K, Endo H, Chan PH (2008) Induction of MMP-9 expresion and endothelial injury by oxidative stress after spinal cord injury. J Neurotrauma 25:184-195.
Yu T, Li Y, Bian A, Zuo H, Zhu T, Ji S, Kong F, Yin D, Wang C, Wang Z, Wang H, Yang Y, Yoo BC, Cho JY (2014) The regulatory role of activating transcription factor 2 in inflammation. Mediators Inflamm 2014:950472.
Yune TY, Lee JY, Jung GY, Kim SJ, Jiang MH, Kim YC, Oh YJ, Markelonis GJ, Oh TH (2007) Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J Neurosci 27:7751-7761.
Zhang H, Chang M, Hansen CN, Basso DM, Noble-Haeusslein LJ (2011) Role of matrix metalloproteinases and therapeutic benefits of their inhibition in spinal cord injury. Neurotherapeutics 8:206-220.
Zhang Y, Chen H, Chen Y, Wang L, Cai Y, Li M, Hui-qin W, Du J, An R, Luo Q, Wang X, Lun Z, Xu Y, Shen J (2014) Activated microglia contribute to neuronal apoptosis in toxoplasmic encephalitis. Parasit Vectors 7:1-11.
Zhao F, Hua Y, He Y, Keep RF, Xi G (2011a) Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke 42:3587-3593.
Zhao P, Waxman SG, Hains BC (2007) Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci 27:2357-2368.
Zhao Z, Liu N, Huang J, Lu PH, Xu XM (2011b) Inhibition of cPLA2 activation by Ginkgo biloba extract protects spinal cord neurons from glutamate excitotoxicity and oxidative stress-induced cell death. J Neurochem 116:1057-1065.
Zhou X, He X, Ren Y (2014) Function of microglia and macrophages in secondary damage after spinal cord injury. Neural Regen Res 9:1787-1795.
Zhu S, Stavrovskaya I, Drozda M, Kim B, Ona V, Li M, Sarang S, Liu A, Hartley D, Wu D, Gullans S, Ferrante R, Przedborski S, Kristal B (2002) Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417:74-78.
Zhu X, Sano H, Kim KP, Sano A, Boetticher E, Munoz NM, Cho W, Leff AR (2001) Role of mitogen-activated protein kinase-mediated cytosolic phospholipase A2 activation in arachidonic acid metabolism in human eosinophils. J Immunol 167:461-468.
*< class="emphasis_italic">Correspondence to: Yinghui Zhong, Ph.D., yz348@drexel.edu.
Yinghui Zhong, Ph.D., yz348@drexel.edu.
orcid: 0000-0002-1363-9466 (Yinghui Zhong)
10.4103/1673-5374.206633
Accepted: 2017-04-28
杂志排行

中国神经再生研究(英文版)的其它文章
- Cerebral mechanism of puncturing at He-Mu point combination for functional dyspepsia: study protocol for a randomized controlled parallel trial
- Genetically modifying transcription factors to promote CNS axon regeneration
- Inhibition and enhancement of neural regeneration by chondroitin sulfate proteoglycans
- Collapsin response mediator protein-2 plays a major protective role in acute axonal degeneration
- Hypoxia inducible factor-1 alpha stabilization for regenerative therapy in traumatic brain injury
- MRI analysis and clinical significance of lower extremity muscle cross-sectional area after spinal cord injury