Therapeutic opportunities and challenges of induced pluripotent stem cells-derived motor neurons for treatment of amyotrophic lateral sclerosis and motor neuron disease
2017-06-05ManojKumarJaiswal
Manoj Kumar Jaiswal
1 Molecular Imaging and Neuropathology Division, New York State Psychiatry Institute, Columbia University, New York, NY, USA
2 Department of Psychiatry, Columbia University, New York, NY, USA
Therapeutic opportunities and challenges of induced pluripotent stem cells-derived motor neurons for treatment of amyotrophic lateral sclerosis and motor neuron disease
Manoj Kumar Jaiswal1,2,*
1 Molecular Imaging and Neuropathology Division, New York State Psychiatry Institute, Columbia University, New York, NY, USA
2 Department of Psychiatry, Columbia University, New York, NY, USA
How to cite this article:Jaiswal MK (2017) Therapeutic opportunities and challenges of induced pluripotent stem cells-derived motor neurons for treatment of amyotrophic lateral sclerosis and motor neuron disease. Neural Regen Res 12(5):723-736.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Amyotrophic lateral sclerosis (ALS) and motor neuron diseases (MNDs) are progressive neurodegenerative diseases that affect nerve cells in the brain affecting upper and lower motor neurons (UMNs/LMNs), brain stem and spinal cord. The clinical phenotype is characterized by loss of motor neurons (MNs), muscular weakness and atrophy eventually leading to paralysis and death due to respiratory failure within 3–5 years after disease onset. No effective treatment or cure is currently available that halts or reverses ALS and MND except FDA approved drug riluzole that only modestly slows the progression of ALS in some patients. Recent advances in human derived induced pluripotent stem cells have made it possible for the first time to obtain substantial amounts of human cells to recapitulatein vitro“disease in dish” and test some of the underlying pathogenetic mechanisms involved in ALS and MNDs. In this review, I discussed the opportunities and challenges of induced pluropotent stem cells-derived motor neurons for treatment of ALS and MND patients with special emphasis on their implications in finding a cure for ALS and MNDs.
iPSCs; stem cells; human patients; ALS; mitochondria; motor neuron disease; disease modeling; neurodegeneration; gene editing; transplantation; drug screening
Introduction
Amyotrophic lateral sclerosis (ALS) and motor neuron disease (MND) are complex neurodegenerative disorders characterized by the selective loss of motor neurons (MNs) of cortex, brain stem and spinal cord leading to progressive weakness with muscle atrophy and eventually result into paralysis and death due to respiratory failure. ALS and MND patients exhibit a wide range of diverse clinical outcomes regarding disease onset, rate of progression and survival (Patten et al., 2014). The occurrence of ALS and MNDs accounts for about 5–7 cases in every 100,000 people, with an addition of ~2 new cases per 100,000 people per year (McDermott and Shaw, 2008). There is currently no satisfactory cure or effective treatment available for ALS and MNDs. Riluzole has been shown to have modest effects on MNs survivalviaexcitotoxicity inhibition (Jaiswal, 2016). ALS and MND patients die usually within 3–5 years after the onset of first symptoms (Gordon, 2013). The majority of ALS cases are sporadic, but ~10% of ALS cases are familial ALS (fALS) (Kiernan et al., 2011). About 20% of these fALS are caused by gene encoding Cu/Zn-superoxide dismutase (SOD1) and are the most common. Mutations in several other genes also cause fALSe.g., autosomal dominant familial MN disease including fALS types 3 and 5; fused in sarcoma (FUS); 6, 7, 8, 9 or ANG gene; 10 or TAR DNA-binding protein 43 (TDP43)/TARDBP, 11 or Figure four gene; NF-H gene; DAO gene; X-linked; chromosome 9 open reading frame 72 (C9orf72), alsin; frontotemporal lobar degeneration (FTLD) and ALS with concomitantFTLD(Adami et al., 2014; Jaiswal, 2014). Likewise spinal muscle atrophy (SMA) caused by genetic mutations on chromosome 5q is the most common MND characterized by the degeneration of alpha MNs (LMNs) in the ventral horns of the spinal cord and MNs in the cranial nerves in the brainstem. SMA is characterized by extreme heterogeneity and is the leading genetic cause of infantile mortality in children (Lorson et al., 2010; Prior et al., 2010). The genetic condition of SMA displays homozygous deletions or mutations of the survival motor neuron (SMN) gene mapped in 5q11.2–q13.3 (Lefebvre et al., 1997). All persons affected by SMA retain a variable number of copies ofSMN2, which correlates to the severity of the disease. The exact functions of the SMN protein are yet to be fully revealed.
Trends
• Cells cannot be manipulated to assume different fates untilits demise, an old dictum falter a decade ago, when Takahashi and Yamanaka (2006) showed that a small number of genes could convert adult fibroblasts into induced pluripotent stem cells (iPSCs).
• During the past decade, it has become clear that cellular makeup is not written in marble stone but can be rewritten by a handful of genes and produce unlimited cells of different lineages and can be utilized for treatment of several neurological diseases including ALS and MNDs.
• Modeling ALS and MNDs in a dish is challenging. Challenges include difficulties in recapitulating aging induced mutations of mitochondria.
• iPSCs derived cell reprogramming is feasiblein vivo, paving the way for future mitochondrial replacement therapy or MNs transplantation using endogenous expression of several Yamanaka/transcription factors.
Significance
• The causes of ALS/MNDs remain largely obscure and current treatments are very limited; there is a pressing need to develop therapeutics drugs based on pathophysiology of ALS and MNDs. iPSCs provide novel approaches to model ‘disease in dish’ for drug discovery with better chances of success and predictability of their effects in humans.
Glossary
• Amyotrophic lateral sclerosis and motor neuron diseases: Neurodegenerative disorders affecting MNs, brain stem and spinal cord resulting in paralysis and eventually death.
• Stem cells: Capacity of undifferentiated cells to self-renew and differentiate into other cell lineages.
• Pluripotent cells: Cells with ability to differentiate into any cell type of an individual.
• Cell reprogramming: Experimental protocol allowing the transformation of differentiated cells into iPSCs through the ectopic expression of transcription factors.
• Differentiation/directed differentiation: Cellular reprogramming of one cell type to other without passing through the pluripotent condition.
• Motor neurons/mitochondrial replacement: Therapeutic strategies based on the replacement of damaged or dead MNs/mitochondria.
• Yamanaka factors: The first original combination of Oct4, Sox2, cMyc and Klf4 transcription factors used for cellular reprogramming.
There are several critical challenges and obstacle for progresses to be made in the field of therapies development such as lack of established models and inaccessibility to the cell type of interest involved in pathophysiology of ALS and MNDs. In order to overcome these issues, iPSC-based technology may represent a valuable option. However, there are several critical challenges of therapeutic translation of induced pluripotent stem cells (iPSCs) in treatment of ALS and MND, namely: absence of clear knowledge regarding the etiopathogenesis and physiopathology of human sporadic/ familial ALS (sALS/fALS) and MND as well as lack of appropriate cell models of the disease to study MNs. Several treatments that have shown promise in transgenic animals with ALS-like disorders failed in human clinical trials (Wijesekera and Leigh, 2009; Bedlack et al., 2015). To develop better therapeutics, researchers needed human MNs but since MNs neither divide nor simply isolated like blood or skin cells from humans, it is a daunting task to isolate enough MNs for drug test. The discovery of iPSCs technique that switchs a skin cells into stem cells (Takahashi and Yamanaka, 2006), when grown in culture renew themselves, can produce an unlimited supply of iPSCs and have the potential to produce cells of every organ type. iPSCs are patient’s specific cells, thus avoiding possible transplantation or graft immunoreactions. Moreover, iPSCs can be produced in substantial amounts, providing an optimal cell source for regenerative therapeutic approaches. Reprogramming technology provides the possibility of differentiating iPSCs towards any cell type of interest which has open the possibility of using skin cells from ALS and MND patients to develop iPSCs, and from them the trillions of MNs and other cell types needed to conduct study for ALS and MND cure (Dimos et al., 2008). These developments signify a great advantage in ALS and MNDs, since obtaining human relevant cells critical for the development ofin vitrodish models that recapitulate mechanisms responsible for the establishment of pathologies, which affect selective MNs of brain stem, spinal cord and cerebral cortex (Chipman et al., 2012; Sandoe and Eggan, 2013).
One of the unsolved mysteries of ALS and MND is why MNs are selectively degenerated and vulnerable to cell death. In recent times, “disease-in-a-dish” models using patient-derived iPSCs as a means to create motor or other neurons and then detect similar alteration in the structure or function of the patient MN is getting momentum for drug discovery and treatment. Although human MNs carrying specific gene mutations have been previously generated from iPSCs, these MNs developed from adult cells harbor specific gene mutations, and thus not relevant to dominant sALS (Di Giorgio et al., 2008; Mitne-Neto et al., 2011). More recently, Alves et al. (2015) made significant progress in isolating fibroblasts from human sALS patients and reprogrammed them into MNs with high yield and established the cell identity using a set of specific markers with a high degree of confidence (Alves et al., 2015). ALS and MNDs with a specific genetic background may gain benefits fromin vitrodish models obtained from iPSC-derived differentiated cells, like iPSC-MNs exhibiting the affected genotype peculiar to the disease (Dimos et al., 2008; Kiskinis et al., 2014). These advantages suggest that iPSCs could be the key to unravel pathogenetic processes behind ALS and MND which are challenging to study in the animal and human models for their specific features (Figure 1). Results obtained employing iPSCs may pave the way to the development of effective treatments targeting specific disease mechanisms. Here, we review the recent advances in the field of iPSCs as regards to their use in modeling and studying ALS and MND pathogenesis.
Modeling ALS and MND using Human iPSCs-Derived Motor Neurons
Human-derived iPSCs platforms have now enabled us for the first time to testin vitrosome of pathogenetic hypotheses of ALS and MND and investigate early disease mechanismsin vitrousing the patients or healthy donor derived cells (Figure 2). Earlier Dimos and colleagues for the first time and recently Kiskinis and colleagues showed that the iPSCs from skin fibroblasts of ALS patients harbored the patient-specific genetic recipe, thus providing a precious tool to model the ALS pathology (Dimos et al., 2008; Kiskinis et al., 2014). They compared patients derived iPSCs differentiating towards MNs with control healthy human iPSCs and found that the differentiated MNs number from patients decreased in culture in a time-dependent manner, whereas non-motor neuronal cells unaffected. Patients derived MNs also showed a detrimental morphology with small fine processes and shrinked soma similar to changes observed in ALS pathophysiology. In addition, zinc finger nuclease (ZFN)-mediated gene correction ofSOD1mutation rescued the altered morphology and reduced lifespan of the cells. Global transcriptomic analysis using RNA-seq ofSOD1MNs revealed strong down-regulation of genes related to mitochondria functions, protein translation and impairment in mitochondria motility. Further investigation also revealed intrinsic hyperexcitability displayed by iPSCs-derived MNs from ALS patients (Sareen et al., 2013; Wainger et al., 2014), correlating ER stress mechanism. Moreover, comparison between iPSCs-derived MNs fromC9orf72andSOD1lines revealed about the altered common pathways related to oxidative stress and decreased mitochondrial activity downstream of these mutations. The aberrant cytoplasmaticTDP43aggregation is another common pathological trait both in fALS and sALS. iPSCs generated from patients carrying theTDP43M337V mutation and healthy controls MNs have no differences in the differentiation and maturation towards a MNs fate but reduced survival, higher levels ofTDP43and altered cytoplasmatic granules motility of cortical neurons (Bilican et al., 2012; Alami et al., 2014). Moreover, comparison of sALS with familial ALS (C9orf72/SOD1/TDP43) would be of interest to identify sALS disease-pathways and genes related to fALS cases.
Mutations in theFUSgene and abnormal aggregation ofFUSprotein have been reported in fALS. Recently fALS-iPSCs fromFUSpatients were established to model for disease and for use in clinical applications. For example, episomal vectors an integration-free iPSCs from peripheral blood mononuclear cells (PBMCs) were harvested from a fALS patient carrying theFUS-P525Lmutation and a healthy control and successfully differentiated into MNs and subsequently detected cytoplasmic mislocalization and formation of FUS protein aggregates in MNs (Liu et al., 2015). Lenzi et al. (2015) successfully differentiated iPSCs derived from FUSR514Sand FUSR521Cpatient fibroblast carrying mutations inFUSinto spinal cord neural cells mimicking the physiological conditions and in the case of the severe FUSP525Lmutation a heterozygous and a homozygous the iPSCc were raised by TALEN-directed mutagenesis due to unavailability of fibroblasts (Lenzi et al., 2015). Japtok et al. (2015) similarly had been successful to differentiate the iPSCs derived from FUS-fALS patient carrying endogenous FUS mutations leading to a benign (R521C) or a more severe clinical phenotype,e.g., frame shift mutation R495QfsX527. Higelin et al. (2016) found that the susceptibility to excitotoxicity was higher in mutatedFUShuman-derived iPSCs and human iPSC-derived MNs than in controls and correlated with the clinical severity of the FUS fALS.
Another hallmark of ALS is neurofilament (NFL) protein aggregation. Using iPSC and transcription activator-like effector nucleases (TALEN) technology to correct theD90ASOD1mutation to avoid concerns related to heterogeneity among individuals, Chen et al. (2014) investigated the causative role of mtSOD1in impairing NFL turnover within MNs in isogenic controls environment. NFL subunits of MNs in ALS developed a tendency of aggregate formation leading to neurite relapse due to the presence of mutatedSOD1, as demonstrated by gain and loss of function experiments.
Until Ebert and Svendsen first established human derived iPSCs recapitulating the MND and SMA pathology (Ebert et al., 2009), only models utilized to study SMA were worms and rodents posing serious limitation in terms of detailed understanding of human pathology in the context of these diseases. Subsequently, iPSCs were generated from two patients affected by SMA type I and from of control human iPSCs-derived MNs (Sareen et al., 2012; Sareen et al., 2013). Patients derived MNs underwent reduction in number and size due to activation of apoptosis pathways compared to the control iPSC-derived MNs confirming the earlier findings (Sareen et al., 2012; Sareen et al., 2013). Sareen and colleague further evaluated MNs degeneration and drug efficacy for iPSC-derived MNs confirming MNs disintegration and inhibition of Fas-receptor mediated apoptotic pathway by administration of a monoclonal antibody targeting the Fas-receptor (Anti Fas-Ab). Antibody targeting the Fas-receptor proved to significantly increase the number of MNs in patients derived SMA and provide a framework for the screening and development of new drug compounds (Figure 2). Corti et al. (2012) utilized viral and transgene-free technique and reprogrammed fibroblasts of type I SMA patients into iPSCs by nucleofecting them with plasmids encoding pluripotency factors. They found reduction in cell size, axonal elongation, resulting in a decreased survival of MNs derived from patient’s iPSCs. Genetic correction ofSMN2using oligodeoxy nucleotides rescues the cellular damage to the defectiveSMN1gene.
Overall, these excitingin vitrodata emphasize the significance of human derived iPSCs and their applicability as valuable tools to investigate and understand the mechanisms involved in the disease pathogenesis to elucidate aspects of ALS and MND multifactorial pathogenesis as well as forhigh-throughput screening of potential therapeutic agents. The refinement of human derived iPSC-based technology platforms can further be utilized forin vitroscreening of thousands of potential therapeutic drugs that previously been identified using rodent models or the agents that are to be tested using human clinical trials (Egawa et al., 2012; Garbes et al., 2013). In nutshell, the prospect of human derived cell type specific reprogramming to generate vulnerable MNs (e.g., HMNs/FMNs), glia, astrocytes or oligodendrocytes specifically damaged in the ALS and MND makes iPSCs an extraordinary tool and can be utilized as a reliable‘disease in dish’ model to test novel therapeutic strategies and unveil pathogenic mechanisms that have been unapproachable so far.
Modeling Physiologically Relevant iPSCs-Derived Motor Neurons with Deadly Neighbors
Though iPSCs-derived MNs somewhat resembled those in people with ALS, one unexplored avenue of disease modeling is to introduce key neighboring instigators such as astrocytes, microglia, oligodendrocyets and endothelial cells into the mix cells to truly recreate the physiologically relevant microenvironment utilizing the iPSCs-derived MNs in laboratory dishes for study. Recently Gage and co-workers found that only half as many MNs survived when exposed to the mutated astrocytes in mix cells, compared with those cultured with the normal gene because mutated astrocytes were producing superoxide which activatedNOX2gene in the neurons implicating the role of oxidative stress of neighboring astrocytes and its implication in impairment of MNs mitochondria (Marchetto et al., 2008). Eggan’s and colleagues found that the human MNs cultured with cortical glia obtained from mtSOD1mice significantly decrease over time suggesting a non-cell autonomous effects and this glial toxicity effect is specific for MNs (Di Giorgio et al., 2008).
Astrocytes were shown to contribute to MN dysfunction in mtSOD1mice model of ALS (Benkler et al., 2013). In order to examine the morphologic and functional abnormalities of astrocytes in MND and SMA, human SMA-derived iPSCs and healthy human-derived iPSCs were differentiated into astrocytes (McGivern et al., 2013). The SMA astrocytes have characteristics of activated phenotype accessed by intense cytoplasmic labelling of glial fibrillary protein (GFAP) and Nestin, the markers of intermediate filament proteins upregulated in reactive glial cells and enlarged bodies and thick, short processes. This observation infers that the MN death is preceded by the activation of glial cells, suggesting a connecting relationship between the two events. Moreover, transplants of human iPSC-derived glial-rich neural progenitors (hiPSC-GRNPs) were able to reduce MN loss and increase lifespan of mtSOD1mice model of ALS (Kondo et al., 2014). When the authors transplanted the hiPSC-GRNPs after the disease onset, improvement of ALS pathology was noted providing the evidence that glial cells play a major role in ALS pathogenesis (Kondo et al., 2014).
iPSCs-Derived Motor Neurons and Mitochondrial Degeneration in ALS and MND
We have known for a long time the link between disrupted mitochondrial homeostasis, dysregulated calcium signaling, and MNs function, distribution and degeneration in ALS and MND (Goos et al., 2007; Jaiswal, 2009, 2013, 2014, 2017; Jaiswal and Keller, 2009). The roles of mitochondrial physiology and mitochondrial genes in ALS and MND have been extensively studied in the mice, cell culture models and patients (Wong et al., 1995; Kong and Xu, 1998; Beal, 2000; Bendotti et al., 2001; Liu et al., 2004; Goos et al., 2007; Di Giorgio et al., 2008; Dimos et al., 2008; Jaiswal and Keller, 2009; Jaiswal et al., 2009, 2012, 2013; Magrané et al., 2013; Parone et al., 2013; Wainger et al., 2014; Paine and Jaiswal, 2016). Recently, Alves et al. (2015) made significant progress in this area in an iPSCs-derived MN model of ALS and identified 105 differentially expressed genes in iPSCs-derived MNs of the sALS patients as compared to the cells derived from healthy controls. Many of these differentially expressed genes were related to the mitochondrion of differentiated MNs from sALS compared to non-ALS samples (mitochondrion part: 65 genes, mitochondrion matrix: 30 genes, mitochondrion lumen: 30 genes). Their work shows that there is a significant pattern of dysregulated genes expression consistent among the 3 lines of iPSCs-derived MNs of sALS. Indeed some of genes for example,C1QBP,CAV2,CTSB,DIABLO,GLRX2,NDUFA4,NLRX1,OPA1,SDHC,SIRT3,SURF1,SDHC,Smac/DIABLO,ATP5B,E2F1,HTRA2,LETM1,LRRK2,POLG,SIRT3,SOD1,VDAC1, thePSMC4,GRN/ C9orf72,ND2,CHCHD10have been correlated to neuronal death, excitotoxic events and oxidative stress in the context of neuronal death in ALS and MND (Alves et al., 2015). In their work, no variations of mitochondrial gene expression within the groups could be noted but a significant difference could be seen between non-ALS and ALS subjects, emphasizing a possible participation of dysregulated mitochondrial genes in the sALS. Large sets of dysregulated gene profiling of the differentiated iPSCs-derived MNs from sALS patients highlight mitochondrial participation in the establishment of autonomous mechanisms associated with sALS. iPSC-derived MNs from ALS patients uncover significant downregulation of gene networks; among them, genes associated with mitochondrial functions are highly enriched, suggesting that metabolic pathways may be of particular importance for ALS and MND and associated neurodegenerative diseases. These above mentioned works and many other recent findings (Dimos et al., 2008; Alves et al., 2015; Lorenz et al., 2017) provide the framework for further characterization of etiology of sALS using iPSCs-derived MND model and shows the efficacy and therapeutic importance of it for treatment of ALS and MND patients (Figure 2).
Box 1: iPSCs offer several advantages over traditional treatment methods
1) Many ethical issues related to stem cells are eliminated by the iPSCs technology.
2) Use of iPSCs may reduce chances of immune rejection due to use of patient specific autologous cells.
3) High-throughput screening for newly developed lead compounds/drugs.
4) Reduced cost and risk of clinical trials.
5) Capability of iPSCs providing consistent phenotypes every time.
6) Correction and targeting of specific loci/sites of genome for alterations of genes of interest.
7) Personalized medicine modeling on a patient-by-patient since iPSCs are derived from individual patients.
Few points are to be noted when interpreting gene expression analysis in a large set of microarray data. In general, regulation of mitochondrial gene expression is not restricted to pathological conditions but instead to a large range of cellular and physiological conditions. Many times there are huge variations of mitochondria mRNA expressions that are not necessarily associated with any pathological transformation. In future, utilizing computational data mining tools to integrate large data sets gathered from patient-derived iPSCs into a comprehensive view of molecular disease events will be helpful for future therapeutics validation.
Role of Advance Gene Editing Technology in iPSCs-Derived Motor Neurons for Cure of Impaired Mitochondria in ALS and MND
Generation of non-contaminated iPSCs has many challenges including incorporation of viral vectors into host genomes, single or multiple vectors, iPSCs source and dependence on factors like usage of small molecules, which make the generation of iPSCs a risky task. We hope in future new generation genome editing tools such as transcription activators like effector nucleases (TALENs), zinc finger nucleases (ZFNs), clustered regularly interspaced short palindromic repeats and CRISPR-associated (CRISPR/Cas) edited reporter iPSCs; cell sorting tools and better fluorescent reporters for the human cell types will solve some of these technical issues and taking disease modeling of iPSCs-derived MNs to new heights.
Attempts have been made recently to combine stem cells and gene therapies for the treatment of ALS and MND related to MNs and mitochondria (Lorenz et al., 2017). They generated human stem cells in the lab, genetically correct iPSCs from patients with mitochondrial DNA disease, repaired common mitochondrial defects, and reported they were able to rescue cell function (Ma et al., 2015; Lorenz et al., 2017). Interestingly, a new paper on stem cell mitochondria points out that iPSCs tend to have more problems if they are derived from older patients and the findings came with two major conclusions, 1) iPSCs clones derived from elderly adults show accumulation of DNA mutations, and 2) acquired mutant DNA mutations can impact metabolic function in iPSCs and might be able to hold back mitochondrial replacement therapies (Kang et al., 2016). However, recent improvement of pronuclear transfer (PNT) technique which solves the problem of mitochondrial carry over and reduces the number of defective mutated mitochondria transferred to the donor cell able to reduce the risk of resurgence of mutant mtDNA (Hyslop et al., 2016). In spite of some strong criticism that iPSCs are incorrectly programmed stem cells because iPSCs reprogramming switches cells into neoplastic metabolism and thus makes them cancerous, experts pursuing mitochondrial replacement believe that mitochondria behave very differently in embryonic stem cells compared to normal human development and levels of mutant mitochondria can fluctuate wildly in stem cells and they seem to be a law unto themselves. Patient-derived iPSCs have been shown to replicate oxidative stress of mitochondria and fALS disease phenotypes (Wainger et al., 2014). An application of TALEN and CRISPR/Cas method to generate iPSC-models of Duchenne muscular dystrophy disease was performed recently (Li et al., 2015). Kiskinis et al. (2014) used ZFN-correctedSOD1mutant iPSCs and revealed a unique, disease-dependent transcriptional signature indicative of increased oxidative stress, reduced mitochondrial metabolism, altered mitochondrial transport using the genome-wide expression analysis of MNs. CRISPR/Cas9 techniques have been combined with advanced bioengineering techniques to create more accuratein vitrodisease models (Wang et al., 2014). Considering these important discoveries, screening cell lines for mitochondrial mutation is especially important if researchers want to use these techniques in clinic. Therefore,in vitroscreening in human iPSCs-derived MNs and reversing associated mitochondrial malfunction may offer invaluable means of understanding intricate molecular signatures of ALS and MND as well as ultimately therapeutic drugs.
Box 2: Clinician’s Corner
1) ALS and MNDs are major neurodegenerative disorders and major health disparities for modern society; treatment to ameliorate MN degeneration will have significant impact on human diseases. iPSCs modeling of ALS and MNDs pathologies provided mechanistic information about pathophysiology of these diseases.
2) MNs/mitochondria reprogramming erases some disease related molecular factors.
3) Identifying the mechanism that controls changes in factors during MNs/mitochondria reprogramming might provide the clues and therapeutic targets for MNs/mitochondria based drug discovery approaches.

Figure 1 Induced pluripotent stem cells (iPSCs) based technology for amyotrophic lateral sclerosis and motor neuron disease modeling and treatment.
The iPSCs-derived MNs data go some way to supportin vivoandin vitroresults in mice demonstrating the role of mitochondria in MNs degeneration. It also prompts a number of predictions: 1) years of research on animal models conclude that animal models alone will not mimics heterogeneous complexity of an ALS patient’s phenotype due to existing variability in the genetic make-up and therefore to mimic the human cell microenvironment synergistic animal- and patient-derived iPSCs technology is way forward 2) in iPSCs, there is no need of proliferation again and again, and, their derivatives are functionalin vitroas well asin vivoafter transplantation and 3) utilizing longitudinal imaging, big data analysis of transcriptomics, proteomics, epigenomics and machine-learning approaches on a large cohort of patient originated iPSCs-derivatives may provide us key ALS-specific mechanistic signatures beyond the phenotype of an individual ALS mutation. The conventional drug discovery study begins with a gene from patients, which is inserted into a mouse, which is then used to test new drugs, which can then be brought back to humans for further testing. With iPSCs, that path is potentially quite different. Skin cells from many different patients can be isolated, converted to stem cells, and used to grow many different lines of MNs. Each can be tested against thousands of drugs in a high-throughput screen to develop early ALS biomarkers enabling faster patient evaluation and diagnosis.
Quality Control in Human iPSCs-Derived Motor Neurons Modeling
An unequivocal and trustworthy method to establish cell lineage is crucial for efficacy of the protocol and interpretation of IPSCs results. Characterization of iPSCs-derived differentiated MNs usingChAT-immunoreactive (IR) and Hb9:GFP reporter labeling (markers of mature MN) as well asOLIG2(oligodendrocyte marker) andPAX6RNAs (transcripts of neuronal progenitor cells (NPCs)) generally carried out and verified with the qPCR in order to confirm that the RNA employed in the microarray corresponded to enriched differentiated MNs. qPCR were generally performed for the analysis and expression of pluripotency markers,NANOG,SOX2,OCT4andChATin the iPSCs and also the expression of thePAX6(neural stem cell (NSC) marker) andOLIG2(MN precursor marker) in the differentiated MNs. Although Hb9::GFP construct is an excellent reporter for MNs, one of the limitations of this techniques is that it does not provide any clue about MNs sub-type identity and occasionally due to long half life of GFP, fluorescence may persist even when Hb9 promoter is no longer active (Li et al., 1998) and provide false MNs lineage identity. Twoin vivomarkers of cell identity,FOXP1-positive cells with LMC identity andHOX5are clinically very relevant and recommended for ALS and MND MNs identification (Amoroso et al., 2013). LMC neurons innervate distal limb muscles and are among the first to die in ALS patients and respiratory failure is the primary cause of mortality of the phrenic and hypaxial motor column, expressingHOX5during differentiation in their mature state (Philippidou et al., 2012). Moreover, it would be necessary to verify whether iPSCs-derived MNs also display the characteristic electrophysiological properties of mature MNs. For example, verification of functional properties of the differentiated MNs and spike frequency adaptationetc. would be interesting to investigate (Manuel et al., 2009). To further verifyin vivofunctionality of mature MNs, experiments must include co-culture of MNS with myotubes as an assay of neuromuscular junction for-mation, a characteristic of ALS and MND pathophysiology. Furthermore, detailed analysis of molecular changes at different phases of MNs in reprogrammed cells is required since ALS is late onset disease. Subtypes of MNs are differently affected in ALS (hypoglossal MNs and facial MNs are susceptible whereas oculomotor MNs and trigeminal MN are resistant), and this selective vulnerability of particular MNs subsets is not well-understood and thus needs to be investigated in future studies employing the iPSCs (Jaiswal, 2012, 2013, 2014).
Box 3: Challenges of iPSCs
1) Reliability to differentiate cells into specific, homogeneous population of neural subtypes devoid of undifferentiated progenitors.
2) Similarity of iPSCs to cancerous cells and its impact on mitochondrial quality control raises safety concerns.
3) iPSCs memory of their origine.g., a NPCs or fibroblast stored in their epigenomes and it have clinical relevance.
4) Mutations of iPSCs and aberrant reprogramming warts of iPSCs epigenome during the cellular reprogramming process and their functional relevance in DNA methylation to unusual patterns of histone modifications.
5) Clinical grade error free human iPSCs-derived differentiated cells is costly affair for fully vetted iPSCs for a specific patient derived from that patient’s somatic cellse.g., skin, fibroblastsetc.
6) Extended time required in culture for maturation and later validation of iPSCs and thus, for most acute and/or life threatening illnesses and injuries, iPSCs just will not be able to be used in a truly patient specific manner.
7) Fetal/juvenile/young/adult “age” of the various neural subtypes not characterized yet.
8) Therapeutic time window for stem cell therapy is not well defined.
9) iPSCs is good for modeling of the disease which is monogenic in nature but not in a situation of complex diseases modeling. 10) Before using iPSCs in clinics it is important to know whether treatment would be autologous or allogeneic.
Transplantation of Human iPSCs-Derived Motor Neurons for Cure of ALS and MND
Several studies in laboratory rodents have shown that transplanted NSCs have remarkable ability to differentiate into brain and spinal cord cells and replace dead cells (Figure 3) (Su et al., 2013). A few clinical trials have also shown that NSCs hold great promise to treat neurological diseases including ALS and MND (Figure 3A–E). Unfortunately, getting NSCs into the spinal cord (SC) or brain stem requires injections directly into the SC by highly skilled neurosurgeons and method is highly invasive and dangerous. Corti et al. (2008) have found an easier way to administered cells. They screened differentiating iPSCs that expressed high levels of aldehyde dehydrogenase, scattered light in a particular way, and expressed the cell adhesion molecule VLA4. Previously the iPSCs that expressed high levels of aldehyde dehydrogenase with low side scattering of light grew well in the spinal cords of rodents, differentiated into nerve cells and relieved symptoms (Corti et al., 2008; Sareen et al., 2014). Transplantation of ALDHhiSSCloVLA4+NSCs labeled with GFP improves neuromuscular function, increases survival and reduces MN and axon loss in ALS mice and no adverse effects, including abnormal cell growth or inflammation, tumor formation in animals (Figure 3A). More recently to determine whether the terminals of iPSC-derived MNs formed synaptic connections with the target muscle, neurofilament (NFL) antibody was used to visualize human iPSC-derived MN axons and rhodamine-conjugated α-bungarotoxin (Yoo et al.) was used to label postsynaptic ACHRs at the motor endplates (Su et al., 2013). Human-specific NFL-positive axons were in close apposition to the motor endplates at 16 weeks after transplantation, whereas no neuromuscular junctions were co-labeled with NFL in vehicle control and normal control rats (Figure 3D and E). Successful electromyography (EMG) responses were induced in all the animals with cell transplants while no EMG response was found in vehicle control animals (Figure 3E). These initial experiments show three possibilities 1) NSCs do not need to be injected into the spinal cord, they can be givenviaintravenous injection and they will enter the SC on their own, a safer mode of administration. 2) NSCs made from iPSCs are apparently do not readily cause tumors and abnormal growth. 3) We do not need fetuses to make therapeutically useful NSC lines rather iPSC technology remove that requirements and ethically more acceptable. Of course, more intense screening is required to establish the safety of iPSCs.
The use of iPSC-derived MNs has a potential for therapeutic uses in MND. In MND for example SMA type I, the production of iPSCs remains a challenge because of the short time window to successfully treat these patients, due to the early loss of MNs. Recent studies where authors reported growth of axons from transplanted MNs can reinnervate the muscle and have functional effects provide hope of generating iPSCs derived from patient with SMA, genetically elevatingSMN1expression, deriving MNs that expressSMN1, and then transplanting these cells back to repopulate and repair the patient’s SC (Figure 3C) (Corti et al., 2008, 2012; Pepper et al., 2016). SMA is a devastating disease and the ability to generate patient-specific iPSCs has opened new avenues of study for cell therapy and disease modeling. iPSC based therapy for SMA may serve as a “proof of concept” that a specific neurologic abnormality can be mirrored in the culture dish.
In summary, MN transplantation raises hope in patients for treating ALS and SMA. However, further research are needed to find the appropriate time window, number of MNs, cocktail of growth factors and other parameters necessary for optimal success of transplantation. Therefore, the ultimate goal for clinician for MN replacement remains the integration, growth, functional maturation, and innervation of muscle. There are several challenges needed to be overcomed but with recent progresses in the iPSCs technology we are moving closer towards therapeutic breakthrough in ALS and MND treatment.
iPSCs-Derived Motor Neurons for Drug Screening
Although mammalian genomes are evolutionarily conserved, humans and mice vary in several aspects of development, particularly during gastrulation and organogenesis. For these reasons, it is preferable to conduct biomedical research in humans, but this is almost always limited toin vitrosystems. Our ability to model complex neurological diseases using human derived iPSCs have revolutionized the ways in which scientists study monogenic, complex, early- and late-onset diseases including brain disorders. Moving a drug from the bench to bedside is very expensive, laborious and slow process. The cost of development and validation of potential therapeutic compounds are enormous. Majority of failures occurred during phase II and III clinical trials of ALS and MND drugs and around 99.99% of drugs in human clinical trials are ultimately not approved for marketing. To date, the only drug commercially available for ALS is Riluzole and its beneficial effects do not exceed more than few months (Cetin et al., 2015; Jaiswal, 2016). The underlying causes of this high rate of failure include the lack of efficacy and clinical safety in patients due to the current limits in disease models in recapitulating the ALS and MND phenotype and in testing drug safety. Instead of treating a ALS and MND patients with consecutive drug therapies until the most suitable one is found, all potential therapeutic drugs and molecules can be tested in parallel on the human-derived patient’s iPSCs with iPSCs from healthy controls facilitating prescription of the most effective drug in human clinical trial. ALS and MND patients and healthy human-derived iPSCs bank consisting of several disease conditions would serve as a source for screening potential drug candidates, predicting the effectiveness of drugs in terms of their dosage, toxicity, and individual patient specific personalized drug response (Giri and Bader, 2015). In this context, human-derived iPSCs provide an exceptional opportunity for drug compound screening as these appropriately mimic the human cells that are affected in the ALS and MND patients. For example, hallmark of ALS disease phenotype includes increased protein aggregate formation and decreased survival rate of selectively vulnerable MNs where asSMN2gene in SMA type I, both of which can be evaluated in iPSCs-derived MNs. Secondly, as a target of screening, iPSCs can be differentiated into the specific MNs subtypes that are most relevant to disease phenotypes, such as selectively vulnerable hypoglossal and facial MNs in ALS. Accordingly, the drugs and therapeutic molecules screened as effective for the given iPSCs-derived MNs are expected to give the similar efficacy when treated to the patients. Furthermore, therapeutic drugs that significantly reverse disease phenotypes and improve deleterious effects in human-derived iPSCs models can then be further tested and modified in order to have better efficacy in ALS and MND patients. An alternative method is to test therapeutic compounds on the basis of our previous understanding of the pathology underlying the ALS and MND cellular phenotypes. For example, recently published results for ALS where they discovered that the patient iPSC-derived MNs displayed hyperexcitability and corrected by a potassium channel agonist that is already FDA-approved for epileptic patients (McNeish et al., 2015). Similar treatments for SMA are also at various stages of clinical trials and hopefully very soon will find their ways to clinic (Sareen et al., 2012; McNeish et al., 2015).
Drug screening for fALS using patient-specific iPSC-derived MNs in human carrying mutations in TAR DNA-binding protein 43 (TDP-43) exhibited shorter neuritis and an increased mutant TDP-43 protein and chemical compounds screening found that a histone acetyltransferase inhibitor Anacardic acid rescued the abnormal ALS MN phenotype (Figure 4A) (Egawa et al., 2012). Patient-derived iPSC-based disease modeling for drug screening of genetically complex sALS using large cohort of healthy controls and ALS patients derived MNs showingde novoTDP-43 aggregation were recently identified and development of small molecule modulators drugse.g., Digoxin and Lanatoside C demonstrated the feasibility of iPSCs utility (Figure 4B) in drug screening (Burkhardt et al., 2013). Ebert et al. (2009) first time used iPSCs to model the specific pathology seen in SMA (Figure 4C–D) and shows it as a promising resource to screen several new therapeutic compounds and drug assays for SMA. Moreover, a new study shown the value of iPSCs where distribution of 55 studies that have used patient-derived iPSCs to successfully treat the aberrant disease phenotype based on classes of genetic disorders, early- and late-onset, screen type and treatment (Figure 4E), and evaluated for drug screening (Avior et al., 2016). To be able to truly fulfill the promise of disease modeling using human-derived iPSCs, we must move from ‘bench to clinic’ and, ideally, find a new treatment for the same ALS and MND patients who donated somatic cells for reprogramming and develop ALS and MND patient-specific personalized therapies.
Conclusions
The pathophysiology of ALS and several MNDs is still elusive, thus obstructing the progress of effective therapies (Gordon, 2013). Several new findings based on human iPSCs-derived MNs in sALS and fALS patients provide new clues about disease pathology (Egawa et al., 2012a; Burkhardt et al., 2013). In SMA, mice lacking the homologousSMN2gene established the clinical symptoms and therefore studies derived from pathological studies need to be validated in human derived cellular models (Schmid and DiDonato, 2007). Human-derived iPSCs-based technology carries the genetic identity of a person, thus permitting the study of patient-specific mutations. Issues related to heterogeneity of iPSCs overcome by a careful experimental protocol whereas cutting-edge molecular methodse.g., TALENs, ZFNs and CRISPR/Cas9 correcting the mutation in patient-derived iPSCs, facilitates obtaining isogenic controls (Corti et al., 2012; Chen et al., 2014). Proper attention needs to be paid to identify cellular subtypes obtained in culture with varying differentiation protocols. Accordingly well-established cellular markers need to be used to assess the neuronal phenotypes as well as the iPSCs maturation condition. Possibility thatreprogrammed cells might maintain epigenetic memory of the cell of origin and thereby interfere with the expression of membrane markers can be avoided by possibility of directly differentiating mature cells (i.e., fibroblasts or astrocytes) into relevant induced cell types (Hermann and Storch, 2013; Vaskova et al., 2013). While these novels new technologies fasten the differentiation protocol, careful long-term observation required from the cell pluripotency to the mature phenotype, in order to monitor time-dependent ALS and MNDs linked alteration.
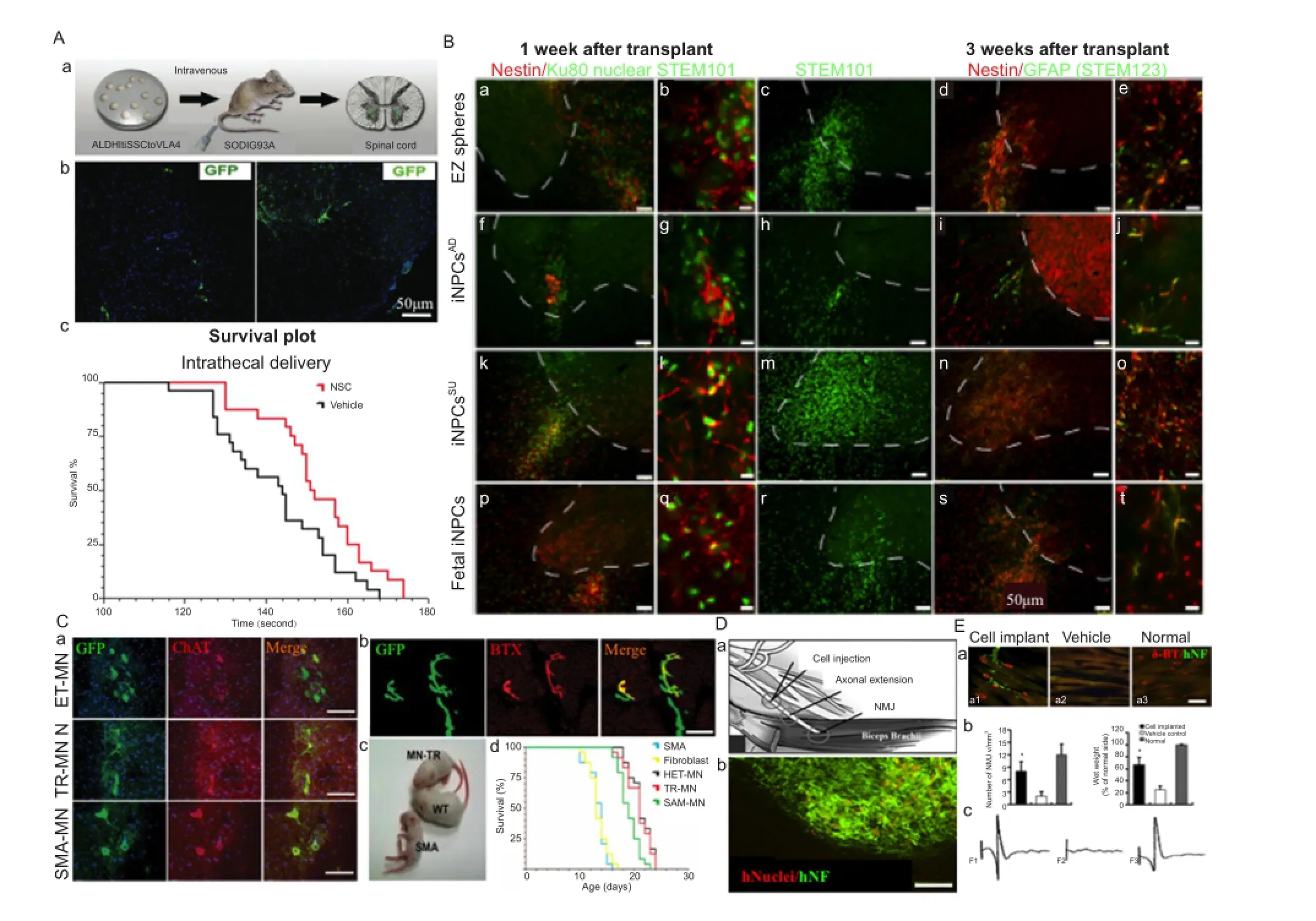
Figure 3 Human induced pluripotent stem cells (iPSCs)-derived motor neurons transplantation in amyotrophic lateral sclerosis and motor neuron disease.

Figure 4 Drug screening using human derived induced pluripotent stem cells (iPSCs).
Outstanding Questions
• What are the best strategies we should developed to recapitulate early and late onset phenotypes in ALS and MNDs experimental ‘disease in dish’ modelsin vitro?
• Do MNs/mitochondria reprogramming erases some disease related molecular factors? Identifying the mechanism that control changes in factors during MNs/mitochondria reprogramming might provide the clues and therapeutic targets for MNs/mitochondria based drug discovery approaches.
• Do specific cell types drive ALS/MNDs disease? Will it possible to restore regenerative capacity of MNs/glia/atrocytes and other cell types vulnerable to ALS/MNDs?
The new development in NSCs field equipped with progress and optimization of iPSC-based technology and advance gene editing has allowed revealing ALS and MND-specific mechanisms, which are neatly patients specific and offers the potential for autologous therapy. While MNs replacement therapy is a future possibility due to successfulin vitroderivation of MNs from iPSCs, these related technologies still need to overcome multiple challenges including unreceptive environment that transplanted cells face in the ALS and MND patients. Original iPSCs programming was carried out by virus transduction and experts cautions about gene activation induced by various viral gene integration or transgene reactivation (Takahashi and Yamanaka, 2006). New programming methodse.g., plasmids, proteins, small molecules and non-integrating viruses should be developed in order to prohibit abnormal reprogramming (Okita et al., 2008; Stadtfeld et al., 2008; Zhou et al., 2009). Finally, while studying the ALS and MND, significance of generation of iPSCs-derived MNs lies not only in new protocol producing differentiated MNs with a higher yield, a higher speed, and a different cell sub-identity, but also in the work of microarray and RNASeq validation and confirmation of up regulation and down regulation of organelles specific genes (e.g., mitochondrial genes) with precision and accuracy, one of the hallmarks of ALS and MNDs. This is potentially a powerful way to find new drug candidates quickly, and to find treatments that may be useful in sALS. With enough patient-derived iPSCs from fibroblast, in near future technology can facilitate ‘in vitroclinical trial’ before a patient-based trial, increasing the likelihood that the therapeutic drugs brought to human patients will work. Disease-in-a-dish modeling takes us a step closer to making iPSCs-modeling of disease a solid technology, offering a reliable alternative to the usual mouse genetic models, and, we hope, eventually delivering on its promise of a new era of individualized medicine for ALS and MND patients and signifies the beginning of a new kind of personalized regenerative medicine. Recent reports provide a glimpse of hope to utilize patient-derived iPSCs for large scale screening of new diagnostic makers, and the perspective of regenerative cellular therapies to improve patient’s quality of life. In summary, it seems that abrupt deleterious changes in selective MNs and mitochondrial and other organelles specific gene regulation sets the stage for MNs degeneration in ALS and MND patients. MNs mitochondria seem to be a key link in the ALS vicious pathophysiological cycle, but other cells may also be involved. These mechanisms can be complementary, and further work is needed to elucidate a potential link between them using iPSCs technology.
Acknowledgments:I would like to thanks Drs. Ananta Paine and Amit Agarwal for comments on the manuscript and valuable discussions.
Author contributions:MKJ conceived of, drafted, edited and approved the manuscript for publication. All aspects of this review article have been solely carried out by MKJ.
Conflicts of interest:None of the authors has any conflict of interest to disclose. The views expressed in this article are those of the author and do not reflect the official policy or position of the NYSPI/Department of Psychiatry, Columbia University Medical center.
Ethical statement:I confirm that I have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Adami R, Scesa G, Bottai D (2014) Stem cell transplantation in neurological diseases: improving effectiveness in animal models. Front Cell Devl Biol 2:17.
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81:536-543.
Alves CJ, Dariolli R, Jorge FMdH, Monteiro MR, Maximino JR, Martins RS, Strauss BE, Krieger JE, Callegaro D, Chadi G (2015) Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front Cell Neurosci 9:289.
Amoroso MW, Croft GF, Williams DJ, O’Keeffe S, Carrasco MA, Davis AR, Roybon L, Oakley DH, Maniatis T, Henderson CE, Wichterle H (2013) Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J Neurosci 33:574-586.
Avior Y, Sagi I, Benvenisty N (2016) Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol 17:170-182.
Beal MF (2000) Mitochondria and the pathogenesis of ALS. Brain 123:1291-1292.
Bedlack RS, Joyce N, Carter GT, Pagononi S, Karam C (2015) Complementary and alternative therapies in ALS. Neurol Clin 33:909-936.
Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, Silani V, De Biasi S (2001) Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci 191:25-33.
Benkler C, Ben-Zur T, Barhum Y, Offen D (2013) Altered astrocytic response to activation in SOD1(G93A) mice and its implications on amyotrophic lateral sclerosis pathogenesis. Glia 61:312-326.
Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, Park IH, Friedman BA, Daley GQ, Wyllie DJ, Hardingham GE, Wilmut I, Finkbeiner S, Maniatis T, Shaw CE, Chandran S (2012) Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A 109:5803-5808.
Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D, Mason M, Garnes J, Dang V, Lievers J, Shoukat-Mumtaz U, Martinez R, Gai H, Blake R, Vaisberg E, Grskovic M, Johnson C, Irion S, Bright J, Cooper B, Nguyen L, et al. (2013) A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci 56:355-364.
Cetin H, Rath J, Fuzi J, Reichardt B, Fulop G, Koppi S, Erdler M, Ransmayr G, Weber J, Neumann K, Hagmann M, Loscher WN, Auff E, Zimprich F (2015) Epidemiology of amyotrophic lateral sclerosis and effect of riluzole on disease course. Neuroepidemiology 44:6-15.
Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, Blackbourn LW 4th, Huang CL, Errigo A, Yin Y, Lu J, Ayala M, Zhang SC (2014) Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14:796-809.
Chipman PH, Toma JS, Rafuse VF (2012) Generation of motor neurons from pluripotent stem cells. Prog Brain Res 201:313-331.
Corti S, Nizzardo M, Simone C, Falcone M, Nardini M, Ronchi D, Donadoni C, Salani S, Riboldi G, Magri F, Menozzi G, Bonaglia C, Rizzo F, Bresolin N, Comi GP (2012) Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci Transl Med 4:165ra162.
Corti S, Nizzardo M, Nardini M, Donadoni C, Salani S, Ronchi D, Saladino F, Bordoni A, Fortunato F, Del Bo R, Papadimitriou D, Locatelli F, Menozzi G, Strazzer S, Bresolin N, Comi GP (2008) Neural stem cell transplantation can ameliorate the phenotype of a mouse model of spinal muscular atrophy. J Clin Invest 118:3316-3330.
Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC (2008) Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3:637-648.
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K (2008) Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321:1218-1221.
Ebert AD, Yu J, Rose FF, Mattis VB, Lorson CL, Thomson JA, Svendsen CN (2009) Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457:277-280.
Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, Aoi T, Watanabe A, Yamada Y, Morizane A, Takahashi J, Ayaki T, Ito H, Yoshikawa K, Yamawaki S, Suzuki S, et al. (2012) Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med 4:145ra104.
Garbes L, Heesen L, Holker I, Bauer T, Schreml J, Zimmermann K, Thoenes M, Walter M, Dimos J, Peitz M, Brustle O, Heller R, Wirth B (2013) VPA response in SMA is suppressed by the fatty acid translocase CD36. Hum Mol Genet 22:398-407.
Giri S, Bader A (2015) A low-cost, high-quality new drug discovery process using patient-derived induced pluripotent stem cells. Drug Discov Today 20:37-49.
Goos M, Zech WD, Jaiswal MK, Balakrishnan S, Ebert S, Mitchell T, Carri MT, Keller BU, Nau R (2007) Expression of a Cu,Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of neuroblastoma cells to infectious injury. BMC Infect Dis 7:131.
Gordon PH (2013) Amyotrophic lateral sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis 4:295-310.
Hermann A, Storch A (2013) Induced neural stem cells (iNSCs) in neurodegenerative diseases. J Neural Transm (Vienna) 120 Suppl 1:S19-25.
Higelin J, Demestre M, Putz S, Delling JP, Jacob C, Lutz AK, Bausinger J, Huber AK, Klingenstein M, Barbi G, Speit G, Huebers A, Weishaupt JH, Hermann A, Liebau S, Ludolph AC, Boeckers TM (2016) FUS mislocalization and vulnerability to DNA damage in ALS patients derived hiPSCs and aging motoneurons. Front Cell Neurosci 10:290.
Hyslop LA, Blakeley 3, Craven L, Richardson J, Fogarty NM, Fragouli E, Lamb M, Wamaitha SE, Prathalingam N, Zhang Q, O’Keefe H, Takeda Y, Arizzi L, Alfarawati S, Tuppen HA, Irving L, Kalleas D, Choudhary M, Wells D, Murdoch AP, et al. (2016) Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 534:383-386.
Jaiswal MK (2012) Motoneuron specific calcium dysregulation and perturbed cellular calcium homestasis in amyotrophic lateral sclerosis: recent advances gained from genetically modified animals and cell culture models. In: Motor neuron diseases: causes, classification and treatments (Turner BJ, Atkin JB eds), pp87-114. New York: Nova Publishers.
Jaiswal MK (2013) Calcium, mitochondria and the pathogenesis of ALS: the good, the Bad and the ugly. Front Cell Neurosci 7:199.
Jaiswal MK (2014) Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol Cell Ther 2:26.
Jaiswal MK (2016) Riluzole but not melatonin ameliorates acute motor neuron degeneration and moderately inhibits SOD1-mediated excitotoxicity induced disrupted mitochondrial Ca2+ signaling in amyotrophic lateral sclerosis. Front Cell Neurosci 10:295.
Jaiswal MK (2017) The role of mitochondria, oxidative stress and altered calcium homeostasis in amyotrophic lateral sclerosis. Lausanne, Switzerland: Frontiers Media.
Jaiswal MK, Keller BU (2009) Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol Pharmacol 75:478-489.
Jaiswal MK, Zech W, Goos M, Leutbecher C, Ferri A, Zippelius A, Carrì MT, Nau R, Keller BU (2009) Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease. BMC Neurosci 10:64.
Japtok J, Lojewski X, Naumann M, Klingenstein M, Reinhardt P, Sterneckert J, Putz S, Demestre M, Boeckers TM, Ludolph AC, Liebau S, Storch A, Hermann A (2015) Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol Dis 82:420-429.
Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y, Koski A, Hayama T, Luo S, Harding CO, Amato P, Jensen J, Battaglia D, Lee D, Wu D, Terzic A, et al. (2016) Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell 18:625-636.
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (2011) Amyotrophic lateral sclerosis. Lancet (London, England) 377:942-955.
Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, Mellin C, Merkle FT, Davis-Dusenbery BN, Ziller M, Oakley D, Ichida J, Di Costanzo S, Atwater N, Maeder ML, Goodwin MJ, et al. (2014) Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14:781-795.
Kondo T, Funayama M, Tsukita K, Hotta A, Yasuda A, Nori S, Kaneko S, Nakamura M, Takahashi R, Okano H, Yamanaka S, Inoue H (2014) Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem Cell Rep 3:242-249.
Kong J, Xu Z (1998) Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci 18:3241-3250.
Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L (2009) Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461:402-406.
Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16:265-269.
Lenzi J, De Santis R, de Turris V, Morlando M, Laneve P, Calvo A, Caliendo V, Chiò A, Rosa A, Bozzoni I (2015) ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis Model Mech 8:755-766.
Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, Yamamoto T, Yamanaka S, Hotta A (2015) Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep 4:143-154.
Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, Kain SR (1998) Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem 273:34970-34975.
Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brännström T, Gredal O, Wong PC, Williams DS, Cleveland DW (2004) Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43:5-17.
Liu X, Chen J, liu W, Li X, Chen Q, Liu T, Gao S, Deng M (2015) The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the FUS-P525L mutation. Neurogenetics 16:223-231.
Lorenz C, Lesimple P, Bukowiecki R, Zink A, Inak G, Mlody B, Singh M, Semtner M, Mah N, Auré K, Leong M, Zabiegalov O, Lyras EM, Pfiffer V, Fauler B, Eichhorst J, Wiesner B, Huebner N, Priller J, Mielke T, et al. (2017) Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell doi:10.1016/j.stem.2016.12.013.
Lorson CL, Rindt H, Shababi M (2010) Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet 19:R111-118.
Ma H, Folmes CD, Wu J, Morey R, Mora-Castilla S, Ocampo A, Ma L, Poulton J, Wang X, Ahmed R, Kang E, Lee Y, Hayama T, Li Y, Van Dyken C, Gutierrez NM, Tippner-Hedges R, Koski A, Mitalipov N, Amato P, et al. (2015) Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 524:234-238.
Magrané J, Cortez C, Gan WB, Manfredi G (2013) Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet 23:1413-1424.
Manuel M, Iglesias C, Donnet M, Leroy F, Heckman CJ, Zytnicki D (2009) Fast kinetics, high-frequency oscillations, and subprimary firing range in adult mouse spinal motoneurons. J Neurosci 29:11246-11256.
Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH (2008) Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3:649-657.
McDermott CJ, Shaw PJ (2008) Diagnosis and management of motor neurone disease. BMJ (Clinical research ed) 336:658-662.
McGivern JV, Patitucci TN, Nord JA, Barabas ME, Stucky CL, Ebert AD (2013) Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 61:1418-1428.
McNeish J, Gardner JP, Wainger BJ, Woolf CJ, Eggan K (2015) From dish to bedside: Lessons learned while translating findings from a stem cell model of disease to a clinical trial. Cell Stem Cell 17:8-10.
Mitne-Neto M, Machado-Costa M, Marchetto MC, Bengtson MH, Joazeiro CA, Tsuda H, Bellen HJ, Silva HC, Oliveira AS, Lazar M, Muotri AR, Zatz M (2011) Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum Mol Genet 20:3642-3652.
Nizzardo M, Simone C, Rizzo F, Ruggieri M, Salani S, Riboldi G, Faravelli I, Zanetta C, Bresolin N, Comi GP, Corti S (2014) Minimally invasive transplantation of iPSC-derived ALDHhiSSCloVLA4+ neural stem cells effectively improves the phenotype of an amyotrophic lateral sclerosis model. Hum Mol Genet 23:342-354.
Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S (2008) Generation of mouse induced pluripotent stem cells without viral vectors. Science 322:949-953.
Paine A, Jaiswal MK (2016) Promise and pitfalls of mitochondrial replacement for prevention and cure of heritable neurodegenerative diseases caused by deleterious mutations in mitochondrial DNA. Front Cell Neurosci 10:219.
Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW (2013) Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci 33:4657-4671.
Patten SA, Armstrong GA, Lissouba A, Kabashi E, Parker JA, Drapeau P (2014) Fishing for causes and cures of motor neuron disorders. Dis Model Mech 7:799-809.
Pepper JP, Wang TV, Hennes V, Sun SY, Ichida JK (2016) Human induced pluripotent stem cell-derived motor neuron transplant for neuromuscular atrophy in a mouse model of sciatic nerve injury. JAMA Facial Plast Surg doi:10.1001/jamafacial.2016.1544.
Philippidou P, Walsh CM, Aubin J, Jeannotte L, Dasen JS (2012) Sustained Hox5 gene activity is required for respiratory motor neuron development. Nat Neurosci 15:1636-1644.
Prior TW, Snyder PJ, Rink BD, Pearl DK, Pyatt RE, Mihal DC, Conlan T, Schmalz B, Montgomery L, Ziegler K, Noonan C, Hashimoto S, Garner S (2010) Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet Part A 152a:1608-1616.
Sandoe J, Eggan K (2013) Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat Neurosci 16:780-789.
Sareen D, Ebert AD, Heins BM, McGivern JV, Ornelas L, Svendsen CN (2012) Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One 7:e39113.
Sareen D, Gowing G, Sahabian A, Staggenborg K, Paradis R, Avalos P, Latter J, Ornelas L, Garcia L, Svendsen CN (2014) Human induced pluripotent stem cells are a novel source of neural progenitor cells (iNPCs) that migrate and integrate in the rodent spinal cord. J Comp Neurol 522:2707-2728.
Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5:208ra149.
Schmid A, DiDonato CJ (2007) Animal models of spinal muscular atrophy. J Child Neurol 22:1004-1012.
Son MJ, Kwon Y, Son MY, Seol B, Choi HS, Ryu SW, Choi C, Cho YS (2015) Mitofusins deficiency elicits mitochondrial metabolic reprogramming to pluripotency. Cell Death Differ 22:1957-1969.
Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K (2008) Induced pluripotent stem cells generated without viral integration. Science 322:945-949.
Su H, Wang L, Cai J, Yuan Q, Yang X, Yao X, Wong WM, Huang W, Li Z, Wan JB, Wang Y, Pei D, So KF, Qin D, Wu W (2013) Transplanted motoneurons derived from human induced pluripotent stem cells form functional connections with target muscle. Stem Cell Res 11:529-539.
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663-676.
Vaskova EA, Stekleneva AE, Medvedev SP, Zakian SM (2013) “Epigenetic memory” phenomenon in induced pluripotent stem cells. Acta Naturae 5:15-21.
Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, Jr., Cudkowicz ME, Bean BP, Eggan K, Woolf CJ (2014) Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 7:1-11.
Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, Geva J, Roberts AE, Ma Q, Ding J, Chen J, Wang DZ, Li K, Wang J, Wanders RJ, Kulik W, et al. (2014) Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med 20:616-623.
Wijesekera LC, Leigh PN (2009) Amyotrophic lateral sclerosis. Orphanet J Rare Dis 4:3.
Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105-1116.
Xu CC, Denton KR, Wang ZB, Zhang X, Li XJ (2016) Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis Models Mech 9:39-49.
Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, Crabtree GR (2011) MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476:228-231.
Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y, Siuzdak G, Scholer HR, Duan L, Ding S (2009) Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4:381-384.
*< class="emphasis_italic">Correspondence to: Manoj Kumar Jaiswal, Ph.D., mj2750@cumc.columbia.edu.
Manoj Kumar Jaiswal, Ph.D., mj2750@cumc.columbia.edu.
orcid: 0000-0003-3702-2492 (Manoj Kumar Jaiswal)
10.4103/1673-5374.206635
Accepted: 2017-04-17
杂志排行

中国神经再生研究(英文版)的其它文章
- Cerebral mechanism of puncturing at He-Mu point combination for functional dyspepsia: study protocol for a randomized controlled parallel trial
- Genetically modifying transcription factors to promote CNS axon regeneration
- Inhibition and enhancement of neural regeneration by chondroitin sulfate proteoglycans
- Collapsin response mediator protein-2 plays a major protective role in acute axonal degeneration
- Hypoxia inducible factor-1 alpha stabilization for regenerative therapy in traumatic brain injury
- Minocycline targets multiple secondary injury mechanisms in traumatic spinal cord injury