X-连锁慢性肉芽肿病CYBB基因突变分析及产前诊断
2017-05-25李淑娟蒋利萍
李淑娟蒋利萍
1. 重庆医科大学附属儿童医院临床免疫研究室(重庆 400014);2.郑州儿童医院(河南郑州 450000)
X-连锁慢性肉芽肿病CYBB基因突变分析及产前诊断
李淑娟2蒋利萍1
1. 重庆医科大学附属儿童医院临床免疫研究室(重庆 400014);2.郑州儿童医院(河南郑州 450000)
目的分析X连锁慢性肉芽肿病(X-CGD)的临床特征及CYBB基因突变。方法回顾分析1例X-CGD患儿的临床资料及其家系的CYBB基因检测结果。结果男性患儿,新生儿期起病,以反复严重的肺部感染为主要表现。患儿无刺激组及脂多糖(LPS)刺激组四唑氮蓝试验(NBT)均为0%,中性粒细胞氧化指数(NOI)为1.15。基因分析显示,患儿CYBB基因第6外显子出现缺失突变(579-582delATTA),由此引起编码序列从189位异亮氨酸(I)发生移码突变,于212位氨基酸提前出现终止密码子(I189fsX212)。患儿母亲及外祖母均为突变基因携带者。患儿母亲下一胎羊水细胞的CYBB基因未发现相同缺失突变。结论基因诊断1例CYBB基因突变X-CGD患者及其家系,产前基因检测可避免X-CGD患儿出生。
X连锁慢性肉芽肿病; CYBB基因; 产前诊断
慢性肉芽肿(chronic granulomatous disease,CGD)是常见吞噬细胞(中性粒细胞、单核细胞、巨噬细胞和嗜酸性粒细胞)功能障碍的一种原发性免疫缺陷病,由于基因突变引起还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶(NADPH,还原型辅酶Ⅱ)的某些成分缺失或减少,致使不能产生超氧根、单态氧和H2O2,其杀伤功能减弱,导致慢性化脓性感染,形成肉芽肿。X连锁慢性肉芽肿病(X-linked chronic granulomatous disease,X-CGD)为CYBB基因突变所致,约占总CGD的2/3[1],少数为常染色体隐形遗传(autosomal recessive chronic granulomatous disease,AR-CGD)。X-CGD病死率高,早期诊断、及时干预对避免此类患儿出生有重大意义。现回顾分析1例X-CGD患儿的临床特征、实验室资料,患儿和家系成员外周血白细胞及异常基因检测结果,以及携带异常基因的患儿母亲下一胎羊水细胞CYBB基因检测结果。
1 临床资料
先证者,男性,7个月10 d,以发热半月入院。入院前半月,患儿出现发热,体温38℃左右,伴咳嗽、流涕、喷嚏,黄色蛋花样便3、4次/d,量中等,无黏液脓血,无寒战、抽搐。予口服蒙脱石,腹泻好转,仍发热,热峰39.5℃,遂予万古霉素治疗(具体不详),疗效差,且咳嗽加重,阵咳伴呛奶,以“败血症”收入重庆医科大学附属儿童医院。患儿系G2P1,出生体质量4.25 kg,足月顺产,无窒息。患儿母亲曾有一次人工流产史。患儿生后混合喂养,生长发育可。已接种卡介苗及乙肝疫苗。出生后16 天,因“新生儿肺炎,败血症,鼻尖疥肿,右侧胸腔积液,室间隔缺损,房间隔缺损”住院治疗。治疗期间血培养示葡萄球菌、真菌、绿脓杆菌阳性,痰培养示真菌、绿脓杆菌阳性,纤维支气管镜检查肺泡灌洗液发现肺炎克雷伯杆菌。无药物过敏史,无手术外伤史。患儿母亲有5位兄弟均于1岁内以“发热”为主要症状夭折(具体不详),1妹体健(图1)。否认有其他家族遗传性疾病。入院体格检查:体温37.5℃,心率130次/min,呼吸58次/min,体质量8 kg,神清,反应可,面色稍苍白;颈及腹股沟可扪及数枚0.8 cm×1.5 cm大小淋巴结,质软,可活动;皮肤无皮疹及瘀斑,弹性可;无鼻翼煽动,三凹征阴性;咽充血,双肺呼吸音粗,可闻及少许中细湿啰音;心音有力,律齐,心前区未闻及杂音;腹软,肝肋下3 cm,脾肋下2 cm,质软、缘锐;四肢暖。实验室检查:血常规白细胞11.40×109/L,淋巴细胞0.38,红细胞3.63×1012/L,血红蛋白80 g/L,中性细胞0.60,血小板262×109/L,C反应蛋白219 g/L;血沉52 mm/ h;血培养及痰培养均阴性,抗酸杆菌+新隐球菌阴性;IgG 8.65 g/L,IgA 0.53 g/L,IgM 1.56 g/L,C3 0.92 g/L,C4 0.24 g/L,IgE 60.0 IU/mL;CD3 68%,CD4 27%,CD8 38%,CD19 22%,CD16+56 8%,CD4:CD8 0.71;四唑氮蓝试验(NBT)示无刺激组及脂多糖(LPS)刺激组均为0。胸片示双肺病变,以右下肺为著,性质待定;右侧少许胸膜病变可能。胸部增强CT示双肺感染性病变,纵隔多发淋巴结肿大,双肺胸膜轻度增厚显影。心脏彩色超声示室间隔缺损(膜周流入道),房间隔缺损(2型),肺动脉高压(轻度)。

图1 家系图谱
因患儿有反复感染病史,NBT为0,为进一步明确诊断,行中性粒细胞呼吸爆发实验。取患儿及其家族成员、健康对照(志愿者)新鲜静脉血100 μL(肝素钠抗凝),标记为对照管和刺激管,每管中加全血50 μL在管底。静置对照管中加入5 μL二氢罗丹明(DHR123,20 μmol/L),刺激管中加入10 μL佛波酯(PMA,200 ng/mL)及5 μL DHR123,混匀,37℃水浴箱中孵育20 min。每管中加2 mL溶血剂,室温避光10 min。2 500 r/min,离心半径8cm,离心5 min,弃上清,加入2 mL PBS洗涤2次,相同方法离心,弃上清,加200 μL PBS用流式细胞仪测定各管中性粒细胞荧光强度,患儿中性粒细胞氧化指数(Neutrophil oxidative index,NOI,NOI=PMA刺激细胞荧光强度/静止对照组细胞荧光强度)为1.15,母亲20.4,正常对照 102,见图2。
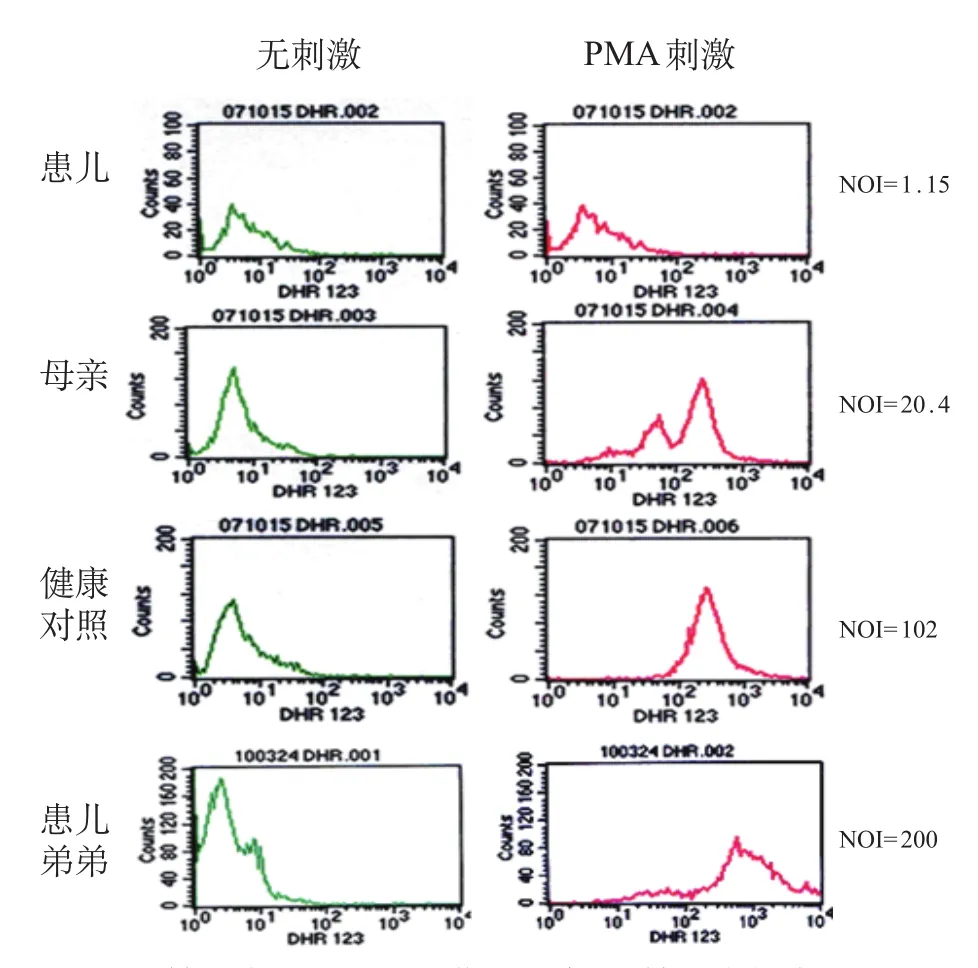
图 2 中性粒细胞呼吸爆发流式图
经重庆医科大学儿童医院伦理委员会审核,家长知情同意后进行基因检测。取患儿、患儿母亲、姨妈及外祖母等家系成员外周抗凝血(肝素钠抗凝)各2 mL,用基因组DNA提取试剂盒(天根公司)提取血细胞和羊水脱落细胞基因组DNA。根据人类基因组数据库(HUGO Gene Nomenclature Committee,HGNC)获得CYBB基因组序列(NC_000023.9)及mRNA序列(NM_000397),应用引物设计软件primer5自行设计引物,由上海生物工程有限公司合成。在20 μL反应体系中进行PCR,10×PCR buffer(Mg2+plus)2.0 μL,2.5 mmol/L dNTP 1 μl,10 μg/L 正、反向引物各 0.3 μL,5 U/μL HS Taq DNA聚合酶 0.3 μL(大连宝生物有限公司TaKaRa产品),70~90 μg/L DNA 2 μL,加双蒸水至 20 μL体积。PCR反应条件:94℃预变性4 min;后按94℃变性30 s,55~63℃退火30 s,72℃ 延伸50 s进行35个循环;最后72℃终末延伸5 min,PCR产物4℃保存。琼脂糖电泳,在紫外透射仪下鉴定扩增结果。扩增产物送至测序中心测序。将测序结果用Bioedit软件与正常序列进行比对,对发现突变的片段重新进行PCR扩增,双向测序,以验证结果的可靠性。先证者CYBB基因第6外显子出现缺失突变(579-582delATTA),引起编码序列从189位异亮氨酸(I)发生移码突变,于212位氨基酸提前出现终止密码子(I189fsX212);患儿母亲外祖母均为突变基因携带者(图3,携带者测序图均为反向测序)。
患儿治疗无效夭折。约2年后母亲再次妊娠,经知情同意后,孕20周时在超声引导下完成羊膜腔穿刺,抽取羊水30 mL至无菌培养管,检测胎儿(先证者弟弟)羊水细胞的CYBB基因,在其外显子6未发现相同缺失突变。胎儿出生后5个月时行呼吸爆发试验,检测NOI为200,示正常。
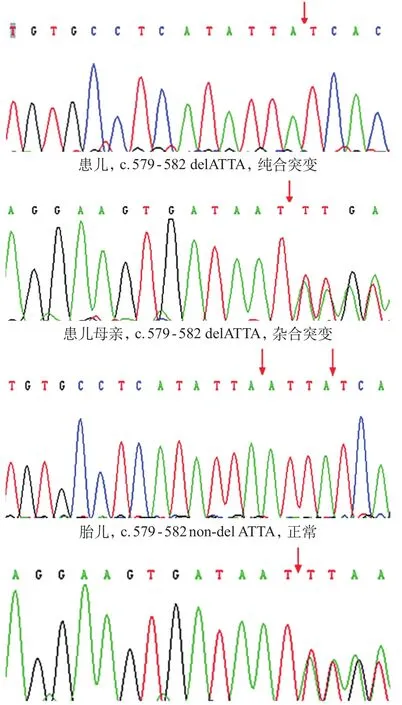
图3 患儿及家系成员CYBB第6外显子基因测序图
2 讨论
CGD是一种少见的原发性吞噬细胞功能缺陷病[2]。X-CGD由于CYBB基因突变导致NADPH氧化酶的β亚单位gp91phox缺陷,不能产生超氧化物,使吞噬细胞杀菌能力减弱或丧失,导致反复严重的细菌和/或真菌感染,以及过度炎症反应形成肉芽肿。美国发病率约为l/(20~25)万[3],我国发病率尚不清楚。
X-CGD为男性患病,女性携带者一般无临床症状,偶见由于X染色体失活所致女性患病报道[4]。反复严重感染是本病的突出表现,肺部病变多见,易感染的病原体分别为曲霉菌、金黄色葡萄球菌及结核分枝杆菌等[5]。在原发性免疫缺陷疾病中,CGD患儿肺部最易合并曲霉菌感染,也是引起患儿死亡的主要原因,患儿可无明显发热、咳嗽等症状,病情恶化时出现高热、咳嗽,甚至咯血,影像学特点是多发结节影及团块影,多为慢性感染,因而空洞及晕轮征较急性感染少见[6,7]。过度活跃的炎症反应形成的肉芽肿为其典型特征,可见于皮肤、胃肠道及泌尿道等,临床上因“胃肠道肉芽肿”引起的顽固性呕吐,易被误诊为“肥厚性幽门狭窄、食物过敏” 等肠道疾病[8],泌尿系肉芽肿引起的血尿也需与急性肾炎、IgA肾病等肾脏疾病相鉴别。本例先证者在新生儿期起病,诊断年龄为7个月左右,首发症状发热,咳嗽,无明显特异性,感染部位以肺部为主,合并症少,无真菌感染的典型肺部影像学表现及明确的病原学支持,白细胞升高不明显,C反应蛋白明显升高;患儿有明显家族史,即母辈中男孩无一存活,尽管免疫缺陷的患儿家族史阳性率不高,但对于反复严重感染的患儿,家族史阳性对于此类疾病的诊断具有提示性。
本例患儿的免疫球蛋白检查及淋巴细胞分类未见明显异常,临床可通过此类检查与严重联合免疫缺陷病、X连锁无丙种球蛋白血症、高IgM综合征及高IgE综合征等其他原发性免疫缺陷病鉴别。
对于反复严重细菌和/或真菌感染并怀疑此病的患儿,中性粒细胞呼吸爆发试验比NBT更快速准确地诊断CGD并发现携带者[9,10],也有检测CYBB基因编码的gp91phox蛋白确诊本病的报道[11]。本例患儿NBT为0,NOI为1.15,其母亲20.4,正常对照102,胎儿出生后5个月龄时的NOI为200,生后一般情况及生长发育均正常。
有报道71例CGD患儿(其中X-CGD53例),起病年龄在23.9个月,平均诊断年龄52.7个月,主要临床表现为反复肺部感染,其次为淋巴结炎、肉芽肿形成,其中30%的患儿存在播散性卡介苗(BCG)病[12]。本课题组此前的研究患儿中,起病年龄平均约在2个月,诊断年龄约2岁,50%的患儿有BCG或结核感染史[13],由此可见CGD患儿易合并结核感染。卡介苗为国家计划免疫疫苗,出生即接种,接种卡介苗后发生结核感染,包括淋巴结、肺、胸膜、肠、脑膜等播散性感染,严重者可致死亡,被称为卡介苗病。对于家族中有可疑免疫缺陷病的新生儿,应加强遗传学咨询,避免接种卡介苗。随着临床医师对本病认识的提高,尤其是采用流式细胞术测中性粒细胞氧化功能后,X-CGD的诊断年龄有所提前,国内曾有新生儿期起病并被确诊为X-CGD的报道[14]。
我国X-CGD发病率尚不明确,如何早期诊断、及时干预,避免此类患儿出生,对优生优育及提高人口质量具有重要意义。本例患儿通过DNA-PCR直接测序法,发现CYBB基因第6外显子存在缺失突变,使编码序列从189位异亮氨酸(I)发生移码突变,于212位氨基酸提前出现终止密码子(I189fsX212),此突变为已报道突变类型,母亲为突变基因携带者,在其下一胎儿胎龄约20周时,通过羊膜腔穿刺抽取羊水,分析羊水脱落细胞的CYBB基因,结果显示正常男性胎儿,于生后5个月时行流式细胞术检测其中性粒细胞氧化指数为200,提示正常,证实产前诊断的准确性。
综上所述,明确先证者的基因突变类型为进一步产前诊断打下基础,而有出生缺陷的阳性家族史为产前诊断提供线索。对于先证者家族中的高风险孕妇,分析其胎儿羊水细胞相关致病基因cDNA,可有效避免CGD患儿出生;致病性基因突变及携带者不能确定而又高度怀疑时,可取高风险孕妇的胎儿脐静脉血行中性粒细胞DHR试验进行产前诊断。
目前X-CGD治疗主要是预防性抗细菌及真菌感染,造血干细胞移植是治愈X-CGD的根本方法[15,16]。早期明确先证者的突变类型、对高危孕妇及时行产前诊断,对预防此类疾病的发生有重要意义。
[1]Ochs HD, Smith CIE, Puck JM. Primary Immunode fi ciency Diseases: A Molecular and Genetic Approach [M]. 2nd Edition. New York: Oxford University Press, 2007:525-545.
[2]Fischer A, Lisowska-Grospierre B, Anderson DC, et al. Leukocyte adhesion de fi ciency: molecular basis and functional consequences [J]. Immunode fi c Rev, 1988, 1(1): 39-54.
[3]Winkelstein JA, Marino MC, Johnston RB Jr, et al. Chronic granulomatous disease.Report on a national registry of 368 patients [J]. Medecine(Baltimore), 2000, 79(3): 155-169.
[4]Lewis EM, Singla M, Sergeant S, et al. X-linked chronic granulomatous disease secondary to skewed X chromosome inactivation in a female with a novel CYBB mutation and late presentation [J]. Clin Immunol, 2008, 129 (2): 372-380.
[5]Mahdaviani SA, Mohajerani SA, Rezaei N, et al. Pulmonary manifestations of chronic granulomatous disease [J]. Expert Rev Clin Immunol, 2013, 9(2): 153-160.
[6]赵成松,赵顺英,刘钢,等.非血液肿瘤和儿科重症监护病房内儿童侵袭性真菌病的高危因素分析[J].中华儿科杂志, 2013, 51(8): 598-601.
[7]Khanna G, Kao SC, Kirby P, et a1. Imaging of chronic granulomatous disease in children [J]. Radiographics, 2005, 25(5): 1183-1195.
[8]Perdereau S, Touzot F, Robin L, et a1. Recurrent pyloric stenosis in a 7-year-old child with chronic granulomatous disease [J]. Arch Pediatr, 2013, 20 (12): 1337-1339.
[9]Ochs HD, Igo RP. The NBT slide test: a simple screening method for detecting chronic granulomatous disease and female carriers [J]. J Pediatr, 1973, 83(1): 77-82.
[10]Alvarez-Larrán A, Toll T, Rives S, et al. Assessment of neutrophil activation in whole blood by fl ow cytometry [J]. Clin Lab Haematol, 2005, 27(1): 41-46.
[11]Sun J, Wang Y, Liu D, et a1. Prenatal diagnosis of x-linkedchronic granulomatousdisease by percutaneous umbilical blood sampling [J]. Scand J Immunol, 2012 , 76 (5): 512-518.
[12]de Oliveira-Junior EB, Zurro NB, Prando C, et a1. Clinical and genotypic spectrum of chronic granulomatous disease in 71 Latin American patients: first report from the LASID Registry [J]. Pediatr Blood Cancer, 2015, 62 (12): 2101-2107.
[13]李淑娟,蒋利萍,刘玮,等.X连锁慢性肉芽肿病12例临床分析[J]. 临床儿科杂志, 2011, 29(1): 42-50.
[14]杨子馨,王亚娟,王帆宁. 新生儿期慢性肉芽肿病6例临床特点及CYBB基因分析[J]. 中华实用儿科临床杂志, 2014, 29(20):1556-1559.
[15]Lin CJ, Wang SC, Ku CL, et a1. Successful unrelated cord blood stem cell transplantation in an X-linked chronic granulomatous disease patient with disseminated BCG-induced infection [J]. Pediatr Neonatol, 2015, 56 (5): 346-350.
[16]Siler U, Paruzynski A, Holtgreve-Grez H, et a1. Successful combination of sequential gene therapy and rescue allo-HSCT in two children with X-CGD - importance of Timing [J]. Curr Gene Ther, 2015, 15 (4): 416-427.
(本文编辑:梁 华)
The analysis CYBB gene mutation and prenatal diagnosis in X-linked chronic granulomatous disease
LI Shujuan2, JIANG Liping1
(1.Clinical Immunology Laboratory, Children's Hospital of Chongqing Medical University, Chongqing 400014, China;2.Children's Hospital of Zhengzhou,Zhengzhou 450000, Henan, China)
ObjectiveTo analyze the clinical feature of X-linked chronic granulomatous disease (X-CGD) and gene mutation of CYBB.MethodThe clinical data of X-CGD in one child and the results of CYBB gene detection in his family were reviewed.ResultsThis boy had onset in the neonatal period and presented with recurrent severe pulmonary infection as his main manifestation.Resultsof nitroblue tetrazolium test (NBT) in both non-stimulation group and LPS stimulation group in the child were 0, and neutrophil oxidation index (NOI) was 1.15. Gene analysis showed a deletion mutation in exon 6 of CYBB gene in the child (579-582delATTA), which resulted in frameshift mutation started from coding sequence of 189—isoleucine (I) and stop codon occured in advance in the 212th amino acid (I189fsX212). Both the child's mother and grandmother were carriers of the mutated gene. The same deletion mutation was not found in the CYBB gene in the amniocyte from the mother's next child.ConclusionOne case of X-CGD patient with CYBB gene mutation and his families were diagnosed by gene detection. Prenatal diagnosis can avoid the birth of children with X-CGD.
X-linked chronic granulomatous disease; CYBB gene; prenatal diagnosis
10.3969/j.issn.1000-3606.2017.04.015
2016-09-05)
