基于竞争性杂交方法的猪-肠道微生物特异性互作靶点发掘
2017-05-19樊丽华莫虹斐张晓峰帅江冰陈青
樊丽华,莫虹斐,张晓峰,帅江冰*,陈青
(1.浙江工商大学食品与生物工程学院,杭州310035;2.浙江省检验检疫科学技术研究院,杭州310016)
基于竞争性杂交方法的猪-肠道微生物特异性互作靶点发掘
樊丽华1,莫虹斐2,张晓峰2,帅江冰2*,陈青11
(1.浙江工商大学食品与生物工程学院,杭州310035;2.浙江省检验检疫科学技术研究院,杭州310016)
动物肠道中存在着一些参与猪肠道微生物互作的基因,这些基因具有一定的宿主特异性,利用其设计分子标记能准确识别粪便污染来源。该文共采集6个物种(猪、牛、羊、鸡、鸭、鹅)的145个粪便样品,提取其DNA后利用竞争性杂交的基因片段富集方法(genome fragment enrichment,GFE),靶向筛选参与猪肠道微生物互作的特异性基因。经BLASTX分析发现,82%的猪特异性非冗余DNA片段存在相似序列,以拟杆菌纲(Bacteroidetes)(43.2%)、梭菌纲(Clostridia)(19.5%)、芽孢杆菌纲(Bacilli)(8.6%)相似序列为主。从蛋白质功能方面分析,61.5%的非冗余序列功能明确,有7.6%的序列与信息贮存及加工有关,12.8%的序列与细胞加工及信息传导有关,22%的序列与代谢有关,其中,碳水化合物和氨基酸的转移代谢相关序列含量最为丰富,均占总特异性序列的6.3%。研究发现,能够编码拟杆菌纲(Bacteroidetes)和梭菌纲(Clostridials)等表面蛋白、膜分泌蛋白及碳水化合物代谢蛋白的相关基因可作为猪特异性分子标记筛选的靶点。
非点源污染;竞争性杂交;特异性基因组片段富集;功能注释;猪特异性分子标记
SummaryDue to the rapid development of livestock breeding and poultry raising,non-point source pollution has become a significant threat to environmental management and aquaculture industry development,as well as to human health in the last few decades.Therefore,it is particularly urgent to establish a monitoring method that can be used as the efficient indicator of fecal pollution with high sensitivity and strong specificity.In animal guts,genes which are directly involved in bacterium-host interactions may display increased level of host-associated genetic variation,making them promising candidates for fecal source tracking.Specific markers targeting bacterium-host interaction genes for human,cattle and chicken were reported previously; however,swine-specific marker for fecal source tracking has not been found yet.
We applied a genome fragment enrichment(GFE)method to enrich swine-specific metagenomic regions that differ from those of other animal species.Briefly,a portion of swine fecal DNA was labeled with biotin and pre-hybridized with a composite fecal DNA pool of other animals including cow(n=20),goat(n=20),chicken(n=8),duck(n=20)and goose(n=5)to block nonunique fragments.Then the pre-hybridized product and another portion of swine fecal DNA labeled with K9 primer were takentogether to perform a competitive DNA hybridization.After streptavidin enrichment and long-linker PCR amplification by K9 primer,the products that were assumed as swine-specific fecal DNA were cloned into vectors and were sequenced.Dot blot hybridization with negative control fecal DNA(composite fecal DNA pool of other animals)was used to identify the cloned GFE sequences which were not swine-specific.The cloned GFE sequences were assigned to bacterial class annotations based on the top BLASTX hit(the lowestE-value score)with the GenBank non-redundant database.The putative protein transcript of each sequence was analyzed based on the similarity of gene sequences by using BLASTX with the GenBank non-redundant database,and their biochemical functions were therefrom predicted.
Sequence analyses of five hundred randomly selected clones from the libraries obtained by three rounds of metagenomic GFE revealed that this subset contained a total of 384 non-redundant sequences and most sequences(87%)ranged from 400 bp to 600 bp in size.Dot blot hybridization using DNA composite of non-target animals as probes showed that only eight clones exhibited cross-reaction,indicating a very low false-positive rate of 2.8%.BLASTX searches identified homologous sequences in GenBank database for 315 non-redundant DNA inserts,with other 69(17.9%of 384 swine fecal DNA sequences)inserts showed no homology with any previously reported genes.Based on top BLASTX hits,the sequences were putatively grouped into 20 bacterial classes including the predominant group of Bacteroidetes-like sequences(43.2%),among which,120 sequences were similar to Bacteroidetes-Prevotella.Clostridia-like sequences were the second most abundant group(19.5%),and Bacillilike sequences represented 8.6%of the clones.Moreover,three sequences exhibited identity to genes in Archaea.Biochemical function annotation revealed that 38.5%of the total analyzed sequences were predicted as genes with unknown functions.Among the fragments associated with characterized function genes(61.5%),the sequences were most frequently assigned to putative proteins associated with metabolism(22%,e.g.,carbohydrate metabolism and amino acid metabolism),cellular process (12.8%,e.g.,membrane transport and DNA repair/replication/recombination)and information storage and processing(7.6%).
It is concluded that gene encoding surface proteins,membrane associated proteins,secretary proteins and carbohydrate metabolism proteins of dominant bacterial classes could be regarded as putative targets for swine-specific microbial genetic markers.
近年来,猪肉制品作为深受国内消费者喜爱的肉制品之一,其庞大的需求量导致生猪产业迅猛发展。据农业部统计,我国生猪出栏量近年来一直占世界第一,占比维持在56%左右,并保持增长态势。由于缺乏对迅猛发展的集约化畜禽养殖业废弃物排放的有效管理,猪、牛、鸡等畜禽粪便及其污水多以非点源的方式进入环境,引发污染并带来健康威胁。猪粪便排泄物中不仅含有大量的病原菌,如沙门菌(Salmonella)、李斯特菌(Listeria)、空肠弯曲菌(Campylobacterspp.)等,而且含有氮、磷等物质,未经有效处理任意排放,一方面会造成土壤地下水、地表水及灌溉水的污染[1];另一方面会使水体富营养化,进而导致水产品蓄积大量的有害物质,影响水产品品质,直接威胁到人类健康。因此,建立一种灵敏度高、特异性强并能高效指示粪便污染源的监测方法显得尤为迫切。
很多研究热衷于从宿主粪便源厌氧菌群如拟杆菌纲(Bacteroidetes)、厚壁菌门(Firmicutes)、双歧杆菌属(Bifidobacteriaspp.)、史氏产甲烷杆菌(Methanobrevibacter smithii)等的相关基因(16S rRNA、毒基因等)筛选特异性标记分子来判别污染来源。目前已鉴定的猪特异性分子标记大多基于猪肠道优势菌拟杆菌16S rRNA的保守区[2-4],也有从史氏产甲烷杆菌中筛选特异性基因mcrA[5],从大肠埃希菌中筛选ST II毒基因[6]及猪肠道腺病毒基因(PAdV)[7]来判断猪粪便污染来源。然而,尽管上述特异性分子标记具有一定的特异性和灵敏性,但也存在较大的局限性:1)宿主特异性不够高,2)不同宿主间存在交叉反应性,3)有地理地域差异性,4)判别动物来源的精确性不够高;因此会带来较高的错判率[8-9]。
有研究者认为,肠道微生物群落与其宿主是共同进化的,经宿主与其肠道微生物之间强烈选择和协同进化可形成宿主微生物相互作用的、对宿主有益的功能保留区(互作靶点),这些互作靶点是形成肠道微生物多样性及特异性的重要因素[10-11]。因此,越来越多的研究从人、牛、鸡等的肠道微生物群落中筛选宿主微生物互作基因作为宿主特异性分子标记,并证明此类分子标记具有较高的特异性和灵敏性[12-14]。然而,目前尚未见以宿主微生物互作基因作为特异性分子标记进行水体或食品中猪粪便非点源污染示踪的相关研究。鉴于此,本研究首次采用竞争性杂交的基因片段富集方法(genome fragment enrichment,GFE)富集猪特异性宏基因组,构建宏基因文库,对文库进行序列菌群分类及功能预测分析,并与不同物种(人、牛、鸡、鼠、鱼、白蚁等)的肠道宏基因组文库结果进行比对分析,从而明确靶向筛选宿主(猪)肠道微生物互作靶点有关的特异性基因,为进一步对猪特异性分子标记设计和猪粪污染源示踪奠定基础。
1 材料与方法
1.1 样品采集和处理
从长江三角洲不同地区8个规模养猪场及其他10个主要的畜禽养殖场(牛、羊、鸡、鸭、鹅)采集新鲜动物粪便样品。将新鲜样品置于无菌容器中,每200 mg(湿质量)样品中加入3 mL GITC缓冲液[5 mol/L异硫氰酸胍、100 mmol/L EDTA(pH 8.0)、0.5%十二烷基肌氨酸钠],低温保存。
共采集6个物种145个粪便样品,其中包括猪粪便样品72个、牛粪便样品20个、羊粪便样品20个、鸡粪便样品8个、鸭粪便样品20个、鹅粪便样品5个。粪样DNA的提取按试剂盒DNA Stool Kit,Qiagen,Valencia,CA,美国)说明书进行,并在ND-2000紫外分光光度计上测定其浓度和纯度(NanoDrop,赛默飞世尔科技公司,美国)。
1.2 宿主(猪)特异性基因组富集(GFE)
将所有单个猪源样品基因组DNA混合形成基因组混合库,以增加基因组的多样性(待富集组),并以牛、羊、鸡、鸭、鹅源样品基因组混合库作为对照组;富集流程见图1。
1.2.1 富集组DNA的准备
将待富集组分为2个组(待富集组A和B)。
A组DNA的准备:将72个纯度在1.8~2.0范围内的猪粪便样DNA各取56 ng混合,声波振荡至150~900 bp,经琼脂糖凝胶电泳鉴定后用7.5 mol/L醋酸铵和100%乙醇沉淀,溶于5 μL TE(10 mmol/L Tris,0.1 mmol/L EDTA,pH 7.5)中,并与4.5 μgK9引物[15](表1)混合放置在95℃下5 min,迅速转至冰上冷却5 min。按照50 U KlenowⅠ聚合酶(新英格兰生物实验室公司)说明书延伸3.5 h。被K9标签标记的DNA片段用试剂盒PCR Purification Kit, Qiagen,Valencia,CA,美国)纯化。利用K9-PCR引物(表1)将纯化产物进行扩增,反应体系100 μL:1×Eaq缓冲液、2.5 mmol/L dNTP、0.2 μmol/LK9-PCR引物、1%乙酰胺、0.6 UEx Taq、K9标记DNA(10 ng)。PCR程序包括:94℃预变性3 min,94℃变性40 s,53℃退火1 min,72℃延伸30 s,共28个循环;最后,在72℃下反应15 min。将5个PCR扩增管内的产物合并,以获得足够量的带有K9随机引物的DNA片段。
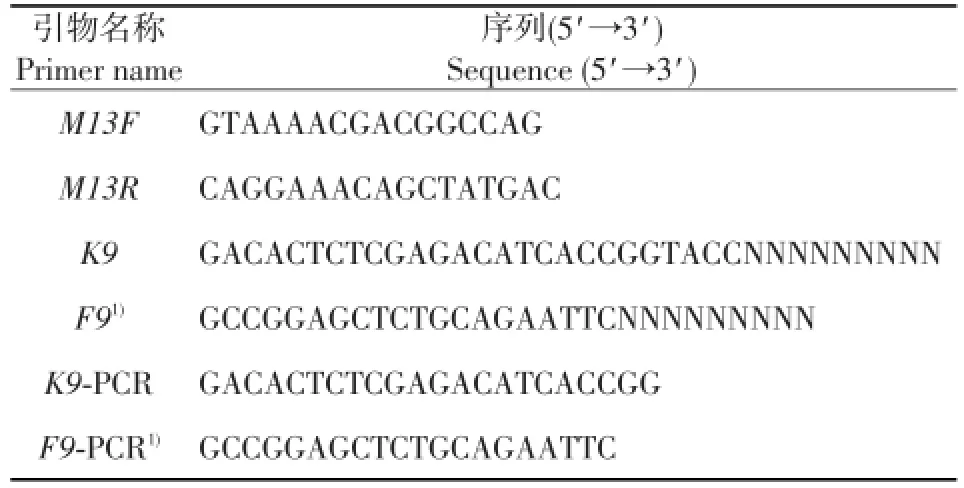
表1 引物序列Table 1 Primer sequences used in the test
B组DNA的准备(生物素标记DNA片段):将72个纯度在1.8~2.0范围内的猪粪便样DNA各取25 ng混合,声波振荡至150~900 bp,经琼脂糖凝胶电泳鉴定后用醋酸铵(7.5 mol/L)和无水乙醇沉淀,并用15 μL TE(pH 7.5)溶解。将溶解后的DNA与1.8 μg生物素(PAB,Sigma-Aldrich,Atlanta,GA,美国)混合,等体积分装至3个PCR微型管(200 μL)中,并将每个PCR微型管放置在距白炽灯(200 W)5 cm下照射20 min,进而生物素化DNA。将以上3个PCR微型管中的DNA混合,经TE(pH 9.0)稀释10倍后用3倍体积的正丁醇萃取以除去未结合的PAB,弃去上清液,将剩余溶液均分成3份,用醋酸铵和乙醇沉淀后溶解于TE中。
1.2.2 对照组DNA的准备
将5种纯度在1.8~2.0范围内的对照组动物粪样DNA分别混合,各取6 μg混匀后声波振荡至150~900 bp,经琼脂糖凝胶电泳鉴定后平均分成3份,经7.5 mol/L醋酸铵和无水乙醇沉淀后用30 μL TE(pH 7.5)溶解。
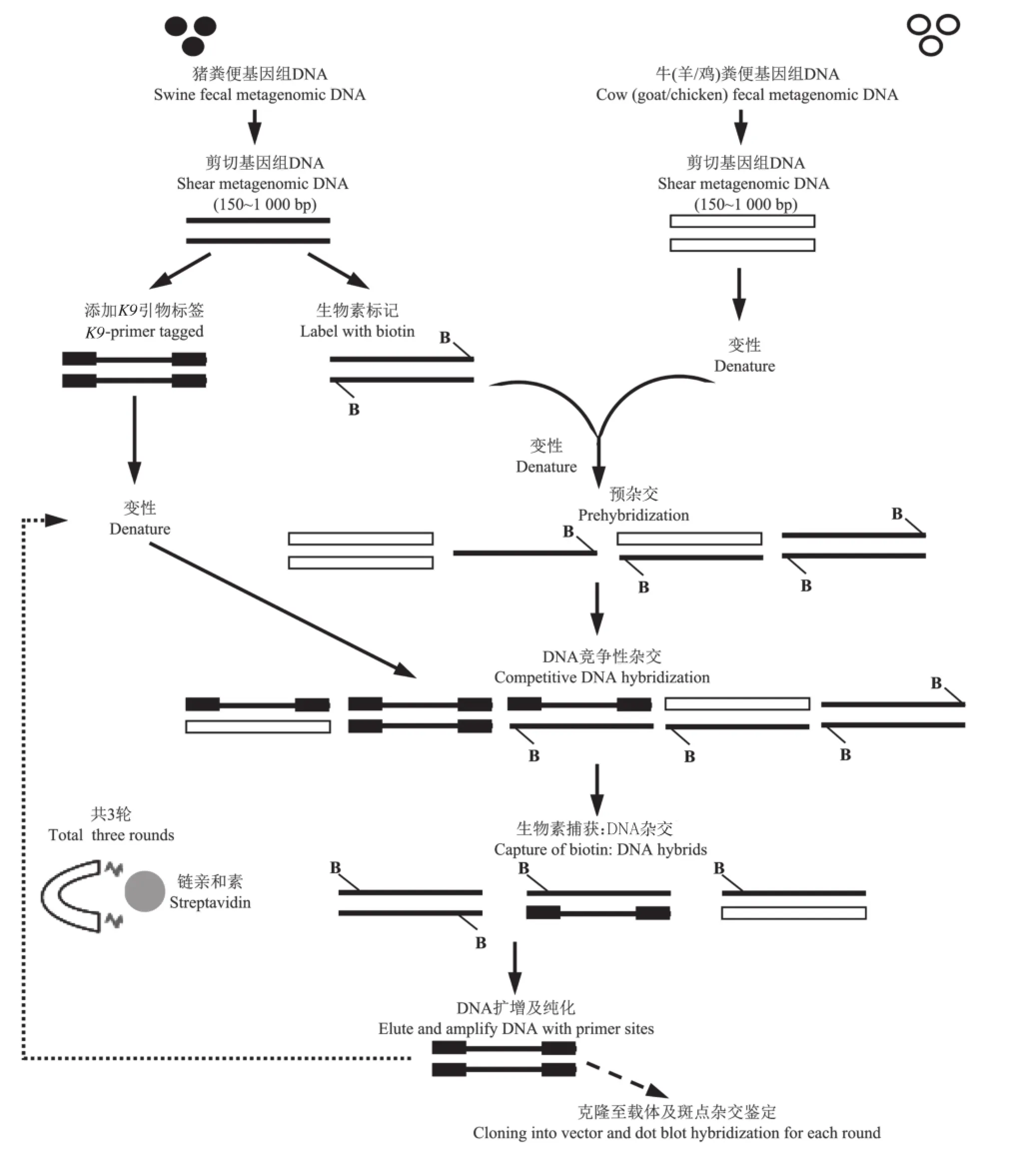
图1 猪粪便特异性基因组富集流程Fig.1 Schematic representation of the DNA enrichment method used to select for swine fecal community DNA sequences
1.2.3 DNA竞争性杂交及富集
将10 μg对照组DNA与0.6 μg B组DNA(生物素标记)混合,经乙醇沉淀后,加入20 μL丙磺酸(EPPS 10 mmol/L,1 mmol/L EDTA),并用矿物质油(Sigma-Aldrich公司,美国)覆盖其表面,在98℃变性2 min,当温度降至55℃时,立即添加5 mol/L NaCl(4 μL),低温杂交30 min。将丙磺酸溶液中的5 μg A组(带有K 9引物)PCR产物经98℃变性2 min后,与已杂交的溶液混合,放置在55℃的恒温箱中过夜进行竞争性杂交。经生物素链亲和素(streptavidin)酶联免疫吸附捕获反应,将标记有生物素的杂交双链DNA(即至少含1条待富集组单链的双链DNA)捕获。利用K9引物扩增所有捕获的杂交双链DNA,扩增条件同1.2.1节。富集过程共3轮,上一轮PCR产物经纯化合并之后用于下一轮的预杂交和杂交,最终获得高丰度的猪源特异性粪样基因组DNA样品。
1.3 非冗余序列分析
1.4 斑点杂交
利用斑点杂交鉴定所富集的序列是否为猪特异性序列。实验方法参见文献[12],简述如下:
1.5 宏基因组文库构建
根据斑点杂交结果,将假阳性率较低的富集轮中的PCR纯化产物克隆至载体pCR TOPO 4.0上,方法步骤同1.3节;挑取阳性克隆送上海生工生物工程股份有限公司进行测序,构建宏基因组文库。
1.6 DNA序列分析
利用DNAStar对所得序列进行拼接及比对。在GenBank数据库中利用BLASTX进行比对分析,根据相似序列的生物功能对每条非冗余序列进行蛋白质功能预测;其中,E值≤1×10-3、识别率≥30%的序列被认为是相似蛋白[16]。通过蛋白相邻类的聚簇(cluster of orthologous groups of proteins,COG)数据库将所得DNA序列进行功能基因分类。根据在GenBank数据库中BLASTX比对的结果(最小E值)对富集的非冗余序列进行菌群分类。
2 结果与分析
2.1 非冗余序列及其假阳性分析
对3轮富集所得的克隆序列进行分析,共获得382个非冗余序列,片段长度为130~924 bp,其中87%为400~600 bp的基因序列。在GFE中第2轮和第3轮所获得的非冗余序列含量较高,而第1轮富集的冗余序列数量较多(表2),其原因可能与克隆增菌培养时间相对较长有关。
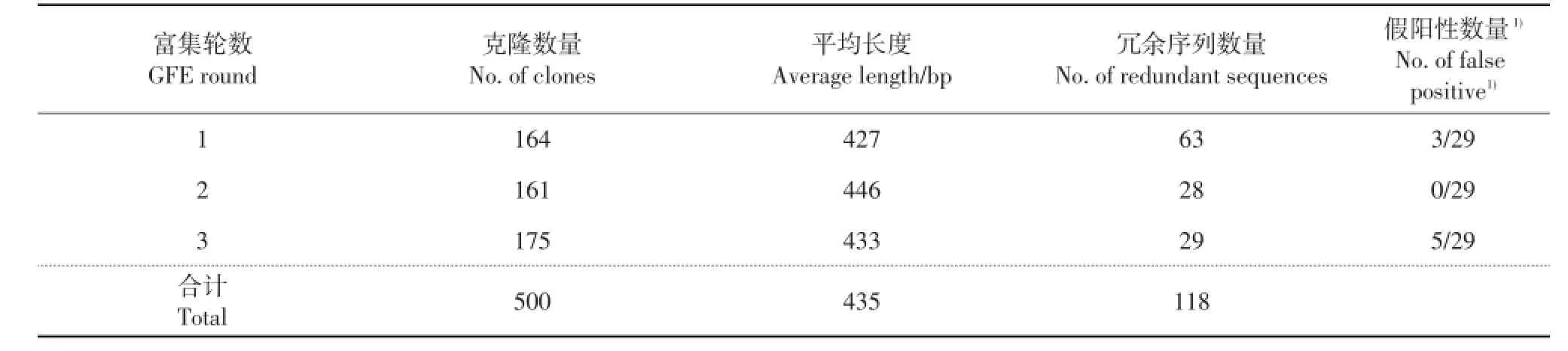
表2 3轮富集克隆序列信息归纳表Table 2 Summary of sequenced DNA clones obtained over three rounds of genome fragment enrichment(GFE)
利用对照组(牛、羊、鸡、鸭、鹅)混合DNA制作探针,从每轮富集的猪非冗余序列中随机挑选29个进行斑点杂交以判断各轮克隆序列的假阳性率(图2)。结果发现,3轮富集只有8个序列能与对照组DNA探针杂交:表明通过竞争性杂交方式富集能获得较低假阳性率(2.8%)的特异性序列。此外,第1轮(n=3)和第2轮(n=0)富集的特异性序列的假阳性率较第3轮低(n=5),且各轮之间假阳性率差异不显著:说明各轮富集的非冗余序列的特异性与富集轮数没有直接相关性。
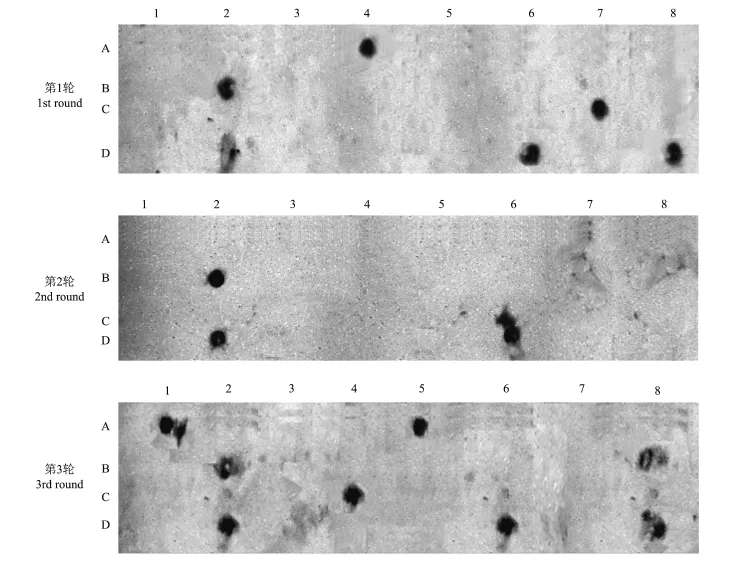
图2 斑点杂交分析所富集的猪特异性DNA片段Fig.2 Dot blot hybridization analysis of putative swine-specific DNA fragments
2.2 猪特异性基因文库菌群多样性分析
挑取假阳性率较低的前2轮富集基因重组菌进行测序分析,共获得384个非冗余猪特异性序列。BLASTX比对结果显示,315个序列(82%)在数据库中具有相似性,而相似序列被编码后共包括19个单元(纲)以及1个古生菌单元(图3)。拟杆菌纲(Bacteroidetes)相似序列含量最多,占43.2%,其中大部分序列(n=120)对普氏菌属(Prevotella)蛋白存在较高的相似性。其次是梭菌纲(Clostridia),占富集的猪特异性基因库序列的19.5%,大多对梭状杆菌属(Clostridials)(3.92%)、罗氏菌属(Roseburia)(3.7%)、毛螺菌属(Lachnospira)(3.7%)中的蛋白表现出相似性。芽孢杆菌纲(Bacilli)相似性序列(8.6%)中多数与乳酸菌属(Lactobacillus)、肠球菌属(Enterococcus)中的蛋白存在相似性。其他序列与来自放线菌纲(Actinobacteria)(1.1%)、变形菌纲(Alphaproteobacteria)(1.1%)、衣原体纲(Chlamydia)(1.3%)等的蛋白相似性较高。此外,还有0.78%的序列与古生菌(Archaea)蛋白相似。上述基于GFE的猪特异性基因文库菌群多样性分析结果与猪肠道微生物的16S rRNA基因组高通量测序结果(图4)相比:拟杆菌纲序列占比基本一致(43.2%与36.8%);但基于GFE的猪特异性基因文库中梭菌纲相似序列的含量显著较低,而芽孢杆菌纲相似序列的含量显著较高。造成这种差异的原因可能与特异性基因在富集过程中引物选择、PCR反应条件及克隆的选择有关[17]。
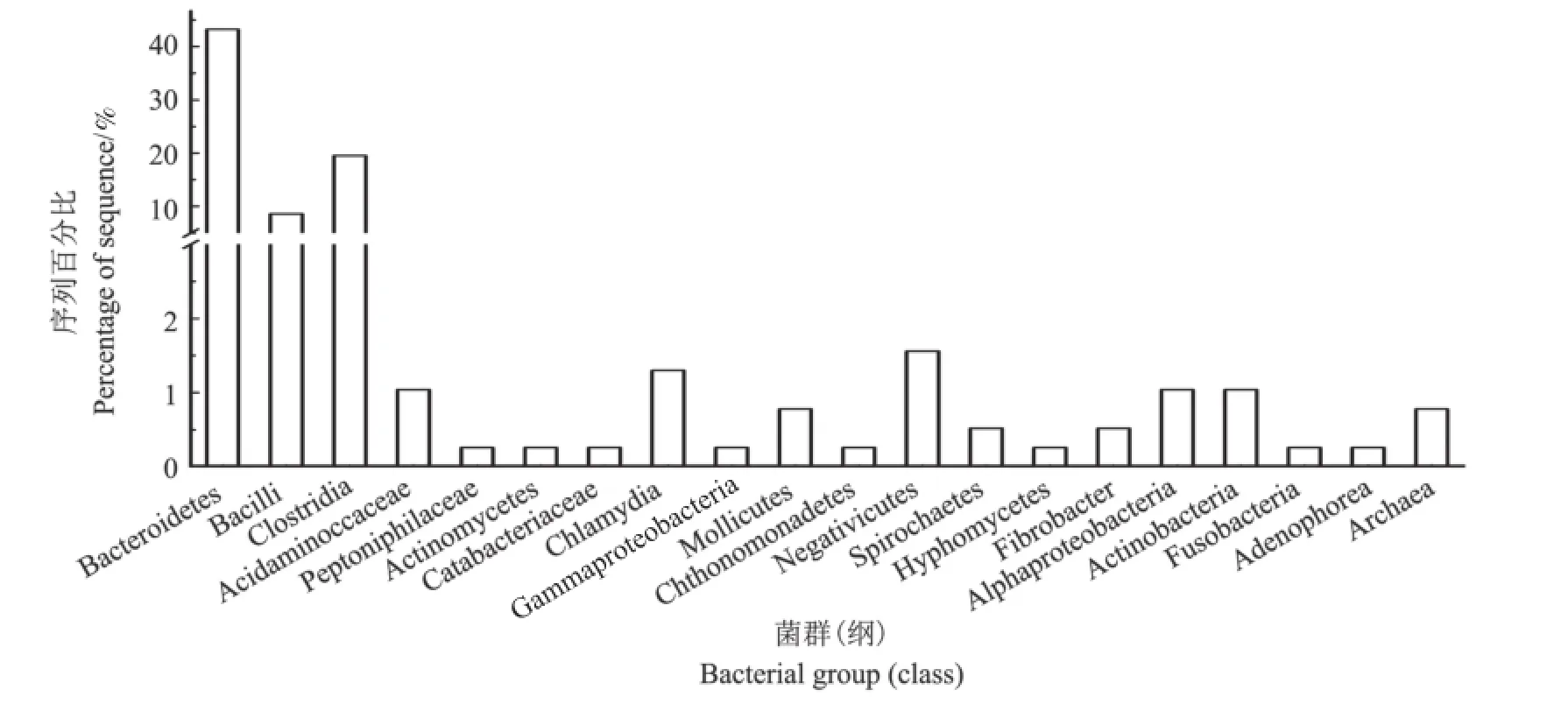
图3 猪特异性DNA片段克隆文库的菌类分析图Fig.3 Bacterial groups associated with the identified swine fecal metagenomic fragments
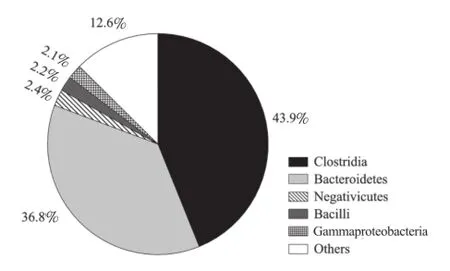
图4 16S rRNA高通量测序在纲水平上分析猪肠道菌群结构Fig.4 Classes of microbial community structure of swine analyzed by high-throughput sequencing
2.3 猪特异性基因文库功能预测分析
经过BLASTX比对分析,根据相似序列的生物功能对每条非冗余序列进行蛋白质功能预测,并通过蛋白相邻类的聚簇(COG)数据库将所得DNA序列进行功能基因分类。结果(图5)显示:在富集的猪特异性基因文库中,功能明确的基因序列占61.5%,其中与拟杆菌纲、梭菌纲相似的序列分别占62.6%、27.2%;29条(7.6%)与信息贮存及加工有关(例如DNA的修复、重组),49条(12.8%)基因序列与细胞加工及信息传导有关,84条(22%)与代谢有关(例如碳水化合物的代谢)。另外有73条(19.1%)与假定蛋白相似。碳水化合物和氨基酸的转移代谢相关序列在功能预测中含量最为丰富,均占总克隆序列的6.3%,这可能与本研究中供试猪摄取高碳水化合物及高蛋白食物有关。其他与细胞膜及细胞壁、DNA复制/重组/修复、保护机制和能量产生及转换的相关基因序列含量也较为丰富。
将本研究中经GFE富集的猪肠道微生物特异性基因文库与近期关于各个物种(人、牛、鸡、鼠、鱼、白蚁等)[18]的肠道微生物宏基因组文库功能预测研究结果进行比对,发现在糖基转移酶系统及碳水化合物代谢系统中一些蛋白(如糖基水解酶、纤维二糖水解酶、麦芽糊精水解、果胶裂解酶等)具有一定的猪特异性。
3 讨论
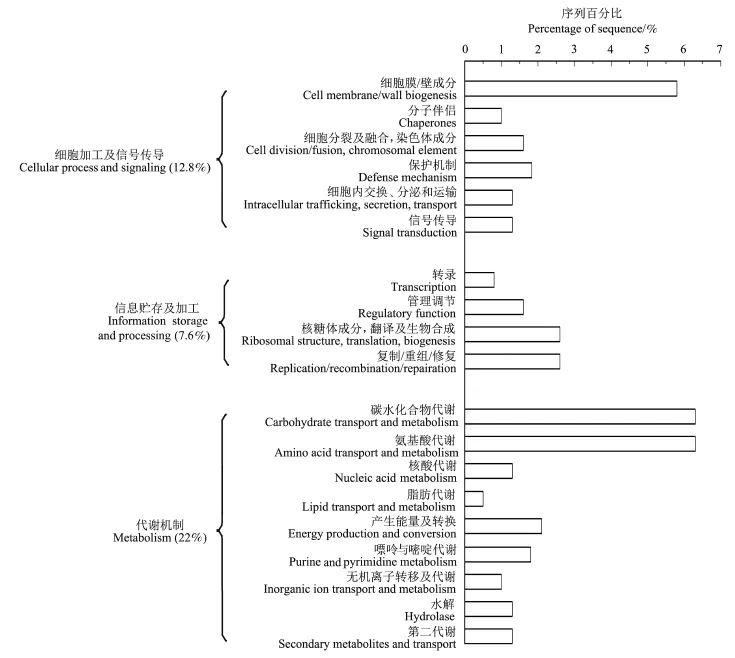
图5 猪肠道特异性克隆序列的功能预测图Fig.5 Function annotation of enriched swine fecal DNA sequences
已有研究表明,拟杆菌纲中具有大量的宿主特异性基因,其中一些参与宿主肠道微生物互作[9,19],例如脆弱拟杆菌(B.fragilis)作为一种革兰氏阴性菌普遍栖息于大多数低血糖哺乳动物中,并具有促进宿主免疫系统细胞的成熟及功能的完善,从而使宿主免受实验性结肠炎威胁的能力[20-21]。梭菌纲也存在参与宿主肠道微生物互作的宿主特异性基因,例如柔嫩梭菌群(Faecalibacterium prausnitzii)作为肠道中最重要的丁酸盐产生菌,它对宿主能量代谢及保持黏膜的完整性影响很大[22]。另有研究证明,每个宿主肠道微生物菌群均有对应的核心菌群和可变菌群,而其含量在个体间存在差异,且造成差异的原因因粪便裂解及DNA提取方法的偏差很难确定,但经过宿主-微生物互作后各个宿主肠道中可变菌群含量普遍较多,且此类菌含有更多的宿主特异性基因[18]。本研究通过2轮生物素链亲和素捕获,将猪基因组中与其他动物基因序列相匹配的去除,从而获得猪特异性序列。从斑点杂交验证性分析可知,GFE是一种能高效率富集并筛选特异性基因的方法。SHANKS等[23]和LU等[14]曾利用竞争性杂交方法分别富集牛和鸡的特异性基因组,结果同样表明经过1轮或者2轮基因组富集就足够获得特异性较高的克隆文库,其特异性与富集轮数未见直接的相关性。经BLASTX分析发现,猪肠道微生物菌群中拟杆菌纲和梭菌纲为优势菌群,可作为宿主肠道微生物互作靶点发掘的目标菌群。此外,对经富集的猪特异性基因库菌群分析表明,古生菌虽含量较低,但是作为共生菌,同样存在着较为密切的宿主微生物互作[9]。例如,作为富集的猪肠道微生物特异性基因库中古生菌的主要组成部分产甲烷古生菌(0.5%),在肠道环境中会消耗氢气进而创造一个有利于发酵型细菌生长的环境,以导致更高能量的摄入。此种现象在肥胖动物群中普遍存在[24],因此,在后续研究中从古生菌类共生菌中挖掘宿主微生物互作基因也是值得探索的。
功能基因组学研究证明,有些功能蛋白如代谢功能蛋白、生理功能蛋白等对宿主具有一定的特异性,即可用于宿主粪便源微生物示踪。目前设计特异性分子标记用于微生物源示踪(microbial source tracking,MST)的功能蛋白主要有微生物表面蛋白(如拟杆菌纲表面的荚膜多糖合成酶、肠球菌表面蛋白esp)、细胞加工(如膜有关分泌蛋白)及一些代谢功能蛋白(如α-1-6甘露聚糖酶、β-葡萄糖醛酸酶)等[12,18,25-26]。将猪特异性基因组文库与牛特异性基因组文库[23]及鸡特异性基因组文库[14]功能预测结果进行比对分析,发现各个功能分类大致相同:说明GFE富集的特异性基因组文库的功能预测种类与实验物种具有相关性,而且各功能序列的相对含量在一定程度上与实验组物种存在一定的联系。将猪与各物种肠道宏基因组蛋白质功能进行比对分析发现,具有猪特异性的蛋白一方面能赋予猪肠道表面多糖结构多样化,使宿主免疫系统适应肠道中微生物的多样性环境;另一方面,使猪肠道摄取利用更多物质,这些特异性蛋白大部分与拟杆菌纲中的蛋白相似,且与宿主微生物互作存在一定的联系[18]。SHANKS等[23]及LU等[14]分别利用拟杆菌纲表面蛋白和参与细胞加工的膜蛋白基因序列设计牛和鸡的特异性分子标记,其MST效果是用16S rRNA作为分子标记所达不到的。鉴于此,在本研究中表面蛋白、膜分泌蛋白及碳水化合物代谢蛋白的相关功能基因可作为猪特异性分子标记研发的靶点。
4 结论
本研究首次利用竞争性杂交的方法富集猪特异性基因,建立了猪肠道微生物特异性基因文库,并结合16S rRNA基因文库和近期各个物种肠道微生物宏基因组文库,对猪肠道微生物特异性基因文库进行菌群分类和功能预测比对分析表明,能够编码优势菌纲(拟杆菌纲和梭菌纲)表面蛋白、膜分泌蛋白及一些碳水化合物代谢蛋白的相关基因可作为猪特异性分子标记筛选的靶点。此研究结果为基于猪特异性分子标记的非依赖型数据库微生物源示踪方法的探索奠定了基础,并对食品和水体安全管理体系和预警体系的发展具有重要意义。进一步工作将在尽量提高富集猪特异性基因方法效率的同时,从猪特异性功能基因中筛选特异性强、灵敏度高的猪特异性分子标签。
[1]CASANOVAS-MASSANA A,GÓMEZ-DOÑATE M,SÁNCHEZ D,et al.Predicting fecal sources in waters with diverse pollution loads using general and molecular host-specific indicators and applying machine learning methods.Journal of Environmental Management,2015,151:317-325.
[2]MIESZKIN S,FURET J P,CORTHIER G,et al.Estimation of pig fecal contamination in a river catchment by real-time PCR using two pig-specific Bacteroidales 16S rRNA genetic markers.Applied&Environmental Microbiology,2009,75(10):3045-3054.
[3]OKABE S,OKAYAMA N,SAVICHTCHEVA O,et al.Quantification of host-specificBacteroides-Prevotella16S rRNA genetic markers for assessment of fecal pollution in freshwater.Applied Microbiology&Biotechnology,2007,74(4):890-901.
[4]DICK L K,BERNHARD A E,BRODEUR T J,et al.Host distributions of uncultivated fecal Bacteroidales bacteria reveal genetic markers for fecal source identification.Applied& Environmental Microbiology,2005,71(6):3184-3191.
[5]UFNAR J A,UFNAR D F,WANG S Y,et al.Development of a swine-specific fecal pollution marker based on host differences in methanogenmcrAgenes.Applied&Environmental Microbiology,2007,73(16):5209-5217.
[6]KHATIB L A,TSAI Y L,OLSON B H.A biomarker for the identification of swine fecal pollution in water,using theST IItoxin gene from enterotoxigenicEscherichia coli.Applied Microbiology&Biotechnology,2003,63(2):231-238.
[7]HUNDESA A,MOTES C M D,ALBINANA-GIMENEZ N,et al.Development of a qPCR assay for the quantification of porcine adenoviruses as an MST tool for swine fecal contamination in the environment.Journal of Virological Methods,2009,158(1/2):130-135.
[8]WITTY M,NICKELS J,LISA J,et al.Ecology,DNA,and the future of microbial source tracking.Water,Air,and Soil Pollution,2009,201(1):219-232.
[9]MCLELLAN S L,EREN A M.Discovering new indicators of fecal pollution.Trends in Microbiology,2014,22(12):697-706.
[10]PANDEYA D R,D'SOUZA R,RAHMAN M M,et al.Hostmicrobial interaction in the mammalian intestine and theirmetabolic role inside.Biomedical Research,2012,23(1):9-21.
[11]VIVEKANANDA M R,RAVINDRA S,SHIVPRASAD D.Oral Host Microbial Interaction.Germany:Lap Lambert Academic Publishing,2015:73-75.
[12]SHANKS O C,DOMINGO J W S,GRAHAM J E.Use of competitive DNA hybridization to identify differences in the genomes of bacteria.Journal of Microbiological Methods,2006,66(2):321-330.
[13]SHANKS O C,DOMINGO J W,LU J,et al.Identification of bacterial DNA markers for the detection of human fecal pollution in water.Applied&Environmental Microbiology,2007,73(8): 2416-2422.
[14]LU J,DOMINGO J S,SHANKS O C.Identification of chickenspecific fecal microbial sequences using a metagenomic approach.Water Research,2007,41(16):3561-3574.
[15]GROTHUES D,CANTOR C R,SMITH C L.PCR amplification of megabase DNA with tagged random primers(T-PCR).Nucleic Acids Research,1993,21(5):1321-1322.
[16]BREITBART M,HEWSON I,FELTS B,et al.Metagenomic analyses of an uncultured viral community from human feces.Journal of Bacteriology,2003,185(20):6220-6223.
[17]CHANDLER D P,FREDRICKSON J K,BROCKMAN F J. Effect of PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries.Molecular Ecology,1997,6(5):475-482.
[18]LAMENDELLA R,DOMINGO J W S,GHOSH S,et al. Comparative fecal metagenomics unveils unique functional capacity of the swine gut.BMC Microbiology,2011,11(10):1-17.
[19]XUB,XUWJ,YANGFY,etal.Metagenomic analysisofthe pygmy loris fecal microbiome reveals unique functional capacity related to metabolismofaromaticcompounds.PLoS One,2013,8(2):118-125.
[20]MAZMANIAN S K,HUA L C,TZIANABOS A O,et al.An immunomodulatory molecule of symbiotic bacteria directs maturation ofthe hostimmune system.Cell,2005,122(1):107-118.
[21]MAZMANIAN S K,ROUND J L,KASPER D L.A microbial symbiosis factor prevents intestinal inflammatory disease.Nature,2008,453(7195):620-625.
[22]PRYDE S E,DUNCAN S H,HOLD G L,et al.The microbiology of butyrate formation in the human colon.FEMS Microbiology Letters,2002,217(2):133-139.
[23]SHANKS O C,DOMINGO J W S,LAMENDELLA R,et al.Competitive metagenomic DNA hybridization identifies hostspecific microbial genetic markers in cow fecal samples.Applied &Environmental Microbiology,2006,72(6):4054-4060.
[24]ZHANG H,DIBAISE J K,ZUCCOLO A,et al.Human gut microbiota in obesity and after gastric bypass.Proceedings of the National Academy of Sciences of the USA,2009,106(7):2365-2370.
[25]XU J,BJURSELL M K,HIMROD J,et al.A genomic view of the human-Bacteroides thetaiotaomicronsymbiosis.Science,2003,299(5615):2074-2076.
[26]RAM J L,RITCHIE R P,FANG J,et al.Sequence-based source tracking ofEscherichia colibased on genetic diversity of betaglucuronidase.Journal of Environmental Quality,2004,33(3): 1024-1032.
Identification of swine-specific microbial genetic markers using competitive DNA hybridization.
FAN Lihua1,MO Hongfei2,ZHANG Xiaofeng2,SHUAI Jiangbing2*,CHEN Qing1(1.School of Food Science and Biotechnology,Zhejiang Gongshang University,Hangzhou 310035,China;2.Zhejiang Academy of Science and Technology for Inspection and Quarantine,Hangzhou 310016,China)
non-point source pollution;competitive DNA hybridization;genome fragment enrichment;function annotation; swine-specific molecular marker
S 182;X 172
A
10.3785/j.issn.1008-9209.2016.06.151
JournalofZhejiang University(Agric.&Life Sci.),2017,43(2):163-172
国家自然科学基金(31301492);浙江省重点研发计划(2015C02044)。
帅江冰(http://orcid.org/0000-0001-6540-1962),Tel:+86-571-81100649,E-mail:sjb@ziq.gov.cn
(First author):樊丽华(http://orcid.org/0000-0002-8534-0914),E-mail:afanlihua@163.com
(Received):2016-06-15;接受日期(Accepted):2016-10-17;
日期(Published online):2017-01-22
猜你喜欢
杂志排行

浙江大学学报(农业与生命科学版)的其它文章
- Evaluation on formation rate of Pleurotus eryngii primordium under different humidity conditions by computer vision
- 流域非点源污染的最佳管理措施成本效益分析研究进展
- Association analysis revealed importance of dominance effects on days to silk of maize nested association mapping(NAM)population
- 检测乙酰微小杆菌的双重实时荧光定量聚合酶链式反应方法的建立
- 应用分子标记辅助选育甘蓝型油菜杂交种的可行性
- 紫萼玉簪种子和幼苗对酸雨与镉复合污染的生理生态响应