3~6月龄伊犁马肠道微生物群落多样性的研究
2017-05-12李晓斌赵国栋吴婷婷何周瑞邓海峰杨开伦新疆农业大学新疆肉乳用草食动物营养实验室乌鲁木齐830052
李晓斌 赵国栋 刘 振 吴婷婷 何周瑞 邓海峰 杨开伦(新疆农业大学,新疆肉乳用草食动物营养实验室,乌鲁木齐830052)
3~6月龄伊犁马肠道微生物群落多样性的研究
李晓斌 赵国栋 刘 振 吴婷婷 何周瑞 邓海峰 杨开伦*
(新疆农业大学,新疆肉乳用草食动物营养实验室,乌鲁木齐830052)
本试验旨在研究3~6月龄伊犁马肠道微生物群落的多样性,揭示断奶前马驹肠道菌群演替的规律,从微生物学角度为此阶段马驹生长发育及肠道健康研究提供理论依据。试验选取平均体重为(89.75±8.81) kg、出生日期相同的3月龄伊犁马5匹,进行为期90 d的饲养试验。分别在试验的第0天(即试验开始的前1天)、第30天、第60天、第90天采集马驹粪便样品,从每份样品中提取微生物基因组总DNA,采用Illumina HiSeq测序技术检测样品微生物群落多样性。结果显示:1)对5匹马驹的20份粪样进行测序,共获得有效序列数157 665条,获得平均操作分类单元(OTUs)1 117个。2)马驹粪便中微生物群落α多样性指数(ACE、Chao1、Shannon和Simpson指数)随着马驹月龄的增加呈波动性变化,但各时间点之间均无显著性变化(P>0.05)。3)在门水平上,马驹粪便中10大优势菌分别为厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、疣微菌门(Verrucomicrobia)、螺旋体菌门(Spirochaetes)、梭杆菌门(Fusobacteria)、无壁菌门(Tenericutes)、放线菌门(Actinobacteria)、TM7、广古菌门(Euryarchaeota),其中以厚壁菌门、拟杆菌门、变形菌门及疣微菌门的丰度较高;在科水平上,马驹粪便中10大优势菌分别为芽孢杆菌科(Bacillaceae)、莫拉氏菌科(Moraxellaceae)、动球菌科(Planococcaceae)、肉杆菌科(Carnobacteriaceae)、BS11、RFP12、乳杆菌科(Lactobacillaceae)、瘤胃球菌科(Ruminococcaceae)、毛螺旋菌科(Lachnospiraceae)、紫单胞菌科(Porphyromonadaceae);在属水平上,马驹粪便中10大优势菌分别为不动杆菌属(Acinetobacter)、德库菌属(Desemzia)、乳杆菌属(Lactobacillus)、Ureibacillus、Paludibacter、芽孢杆菌属(Bacillus)、埃希式杆菌属(Escherichia)、肉食杆菌属(Carnobacterium)、密螺旋体属(Treponema)、艰难杆菌属(Mogibacterium)。由此得出,采用Illumina HiSeq测序技术能够准确地对3~6月龄伊犁马肠道微生物群落进行分类研究;厚壁菌门、拟杆菌门、变形菌门、疣微菌门是3~6月龄伊犁马肠道内的优势菌门。
伊犁马;Illumina HiSeq测序技术;肠道微生物群落;多样性
马是单胃动物,其盲肠相当于反刍动物的瘤胃,可容纳28~36 L的消化物和消化液。通过盲肠内容物或粪便培养检测发现,其微生物种类有400~500种,数量可达1010~1012个/g[1-2]。定植于肠道内的微生物参与肠道营养物质的消化、吸收及合成,是动物肠道重要的组成部分。因此,促进马匹肠道微生物区系的建立,维持肠道正常菌群的平衡对马匹消化吸收、肠道健康和免疫调控意义重大[3]。马驹出生时肠道是无菌的,在分娩、哺乳及与周围环境接触过程逐渐感染细菌。随着马驹年龄、生活环境、饲粮结构、健康状况等的改变,肠道微生物的种类、结构、丰度随之发生变化[4]。在应激条件下这些因素的改变会导致马驹肠道微生物区系紊乱,并引起腹泻,严重时甚至死亡。肠道菌群的平衡在马的生长发育、健康状况方面扮演着重要角色[5]。早期补饲对马驹生长发育意义重大,过早的摄入补喂料对马驹肠道微生物结构改变产生一定的影响。3月龄马驹随着母乳减少逐渐开始增加补喂料,饲粮由液态逐渐转为固态,其肠道微生物的种类和丰度也随之发生变化,从而影响马驹肠道健康和生长发育。鉴于此,本试验以3~6月龄的伊犁马为研究对象,通过检测粪便中微生物群落的多样性,研究马驹从3月龄到6月龄内肠道微生物定植、演替的规律,为研究开发适宜马驹健康的微生物制剂或肠道微生物疾病防治等提供参考依据。
1 材料与方法
1.1 试验时间及地点
试验时间:2014年7月至2014年10月;试验地点:新疆伊犁哈萨克自治州昭苏马场。
1.2 试验设计
本试验选取平均体重为(89.75±8.81) kg、出生日期相同的3月龄伊犁马哺乳马驹5匹。5匹马驹与其母马来自同一放牧草场,且母马年龄(11岁)、胎次(7胎)均相同。在哺乳条件下,每天每匹马驹按照体重的0.6%补喂精料补充料(每30 d调整1次补喂量,具体见表1),并投喂等量的优质苜蓿干草,进行为期90 d的饲养试验。分别在试验的第0天(即试验开始的前1天)、第30天、第60天、第90天12:00前采集马驹粪便样品。
1.3 饲养管理及饲粮组成
试验马驹与母马均饲养于室外马舍(长75 m×宽40 m)。马驹08:00—18:00拴系饲养,其余时间自由活动。将马驹全天的精料补充料分成3等份,分别在08:00、13:00、17:00使用料兜(料兜由本试验设计,其大小和深浅符合马驹采食精料,确保不影响马驹呼吸及防止马驹仰头将精料撒出)补喂。所有试验马驹每天提供等量[(1.00±0.25) kg]的优质苜蓿干草,自由饮水。样品采集当天马驹室内圈舍饲养,08:00将马驹牵入室内圈舍,单槽单栏位饲养,防止马驹粪便互相混合。饲喂后打扫圈舍,为12:00前采集粪样做准备。精料补充料组成及营养水平见表2。
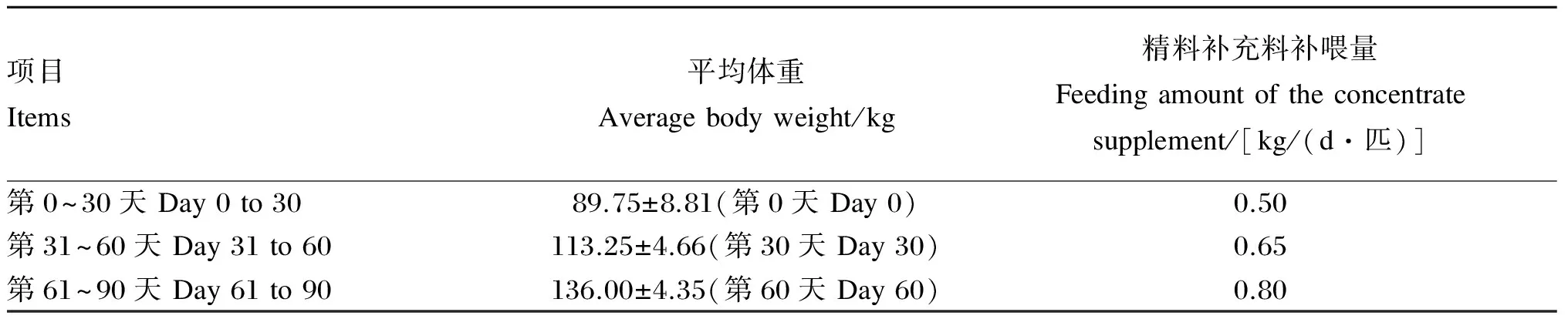
表1 精料补充料补喂量
1.4 马驹粪便样品采集
于试验的第0天、第30天、第60天、第90天的08:00将马驹牵入室内圈舍,单栏位拴系并套带收粪袋。将每匹马驹自进圈后至12:00排出的全部粪便收集于集粪桶内,混合均匀后,采用5点取样法采集马驹粪样,每个点采集5 g,再将5个点共25 g粪样混合均匀后立即装入密封袋,-20 ℃冷冻保存,待测。
1.5 微生物测定样品分组编号
在后续样品测定及分析中分别将试验的第0天、第30天、第60天、第90天的样品记为4个组,每个时间点5匹马驹的样品视为1个组。
1.6 粪样高通量测序分析
总DNA提取、PCR扩增及Illumina HiSeq测序及结果分析均由北京诺禾致源生物信息科技有限公司协助完成。
1.7 数据处理
Metastats分析使用R软件在各分类水平下做组间的置换检验(permutation test),得到P值,然后利用Benjamini and Hochberg False Discovery Rate方法对于P值进行修正。组间差异显著的物种分析利用R软件做组间t检验(t-test)。
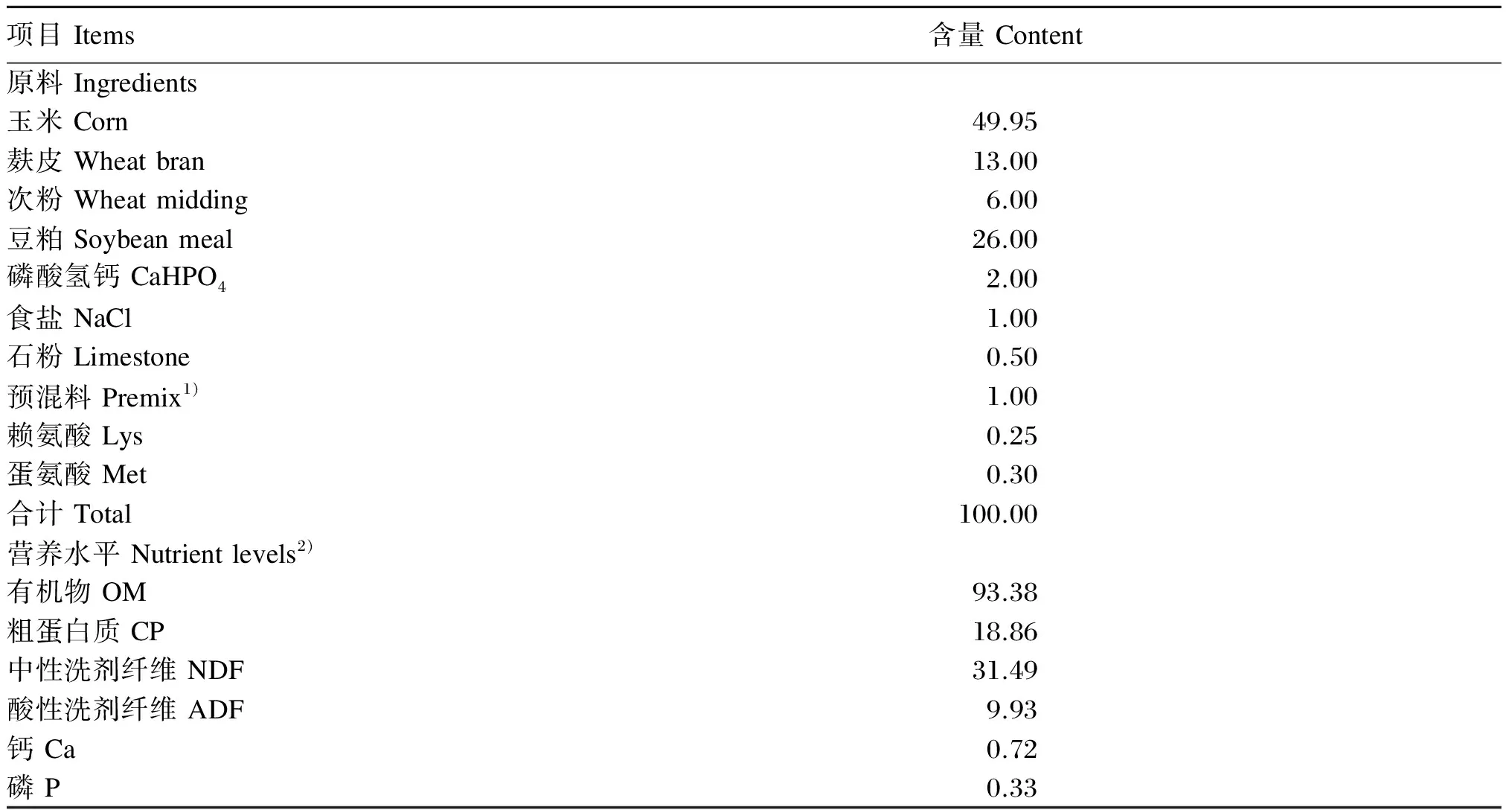
表2 精料补充料组成及营养水平(干物质基础)
1)预混料为每千克精料补充料提供The premix provided the following per kg of the concentrate supplement:VA 3 000 IU,VB120 mg,VB220 mg,VB66 mg,VC 20 mg,VD 1 000 IU,VE 500 IU,泛酸 pantothenic acid 10 mg,烟酰胺 nicotinamide 100 mg,Cu 25 mg,Fe 107 mg,Mn 81 mg,Zn 74 mg,I 6 mg,Se 14 mg,Co 3 mg,氯化胆碱 choline chloride 120 mg。
2)营养水平为实测值。Nutrient levels were measured values.
2 结果与分析
2.1 测序结果及α多样性分析
测序结果及α多样性数据见表3。对5匹马驹的20份粪样进行测序,共获得有效序列数157 665条,以97%的序列一致性(identity),将序列共聚类成4 471个操作分类单元(OTUs)。4个时间点的测序覆盖率均达到96%以上,说明本试验中获得的序列代表了微生物界96%以上的细菌种系型,测序覆盖率好,且各时间点均观察到较丰富的物种。马驹粪便中微生物群落α多样性指数(ACE、Chao1、Shannon和Simpson指数)随着马驹月龄的增加呈波动性变化,但各时间点之间均无显著性差异(P>0.05)。
2.2 β多样性分析
基于OTUs水平的主成分分析(PCA)结果见图1。图中主成分1(PC1)为第1主坐标,对检测到的总微生物的代表性贡献率为17.28%,主成分2(PC2)为第2主坐标,贡献率为9.68%;4个组20个样品能够较明显的区分开,第0天、第30天能够明显与第60天、第90天分开,第60天与第90天能够明显分开,说明同一时间点微生物群落的组成相似。
图2左侧显示的是非加权组平均法(UPGMA)聚类树结构,右侧显示的是各组在门水平上的物种相对丰度分布。可以看出,第0天、第30天聚为一类,第60天、第90天聚为一类,说明随着马驹月龄的增加,微生物群落趋于复杂化和多样化。
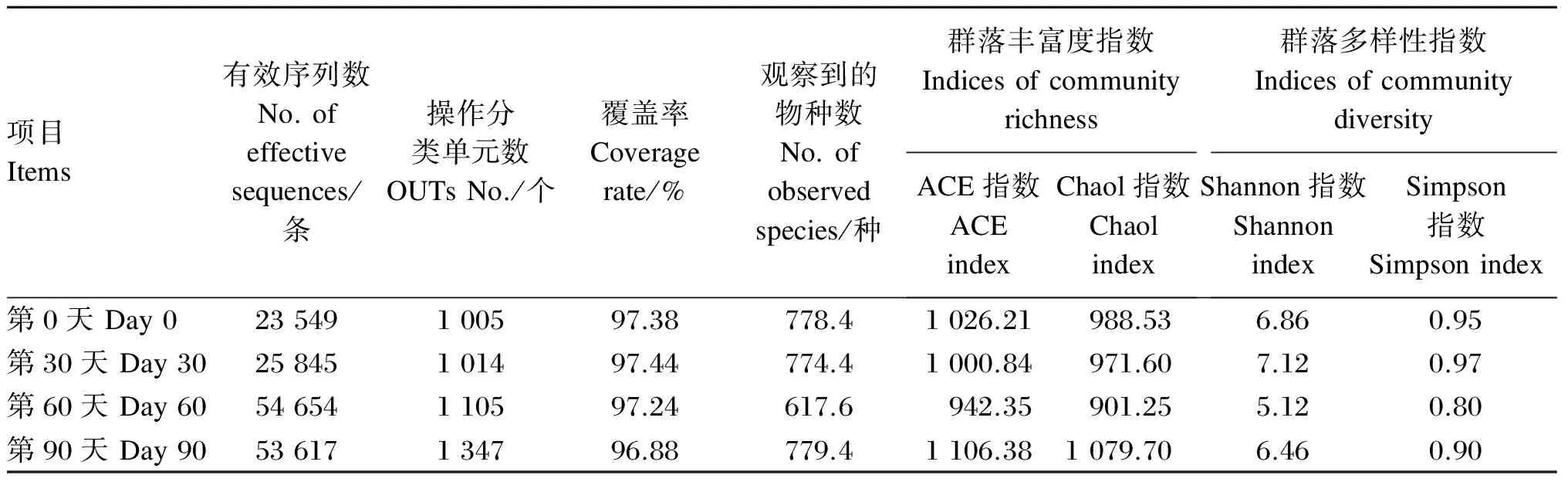
表3 测序结果及α多样性分析
同列数据肩标无字母或相同字母表示差异不显著(P>0.05),不同小写字母表示差异显著(P<0.05),不同大写字母表示差异极显著(P<0.01)。
In the same column, values with no letter or the same letter superscripts mean no significant difference (P>0.05), while with different small letter superscripts mean significant difference (P<0.05), and with different capital letter superscripts mean significant difference (P<0.01).
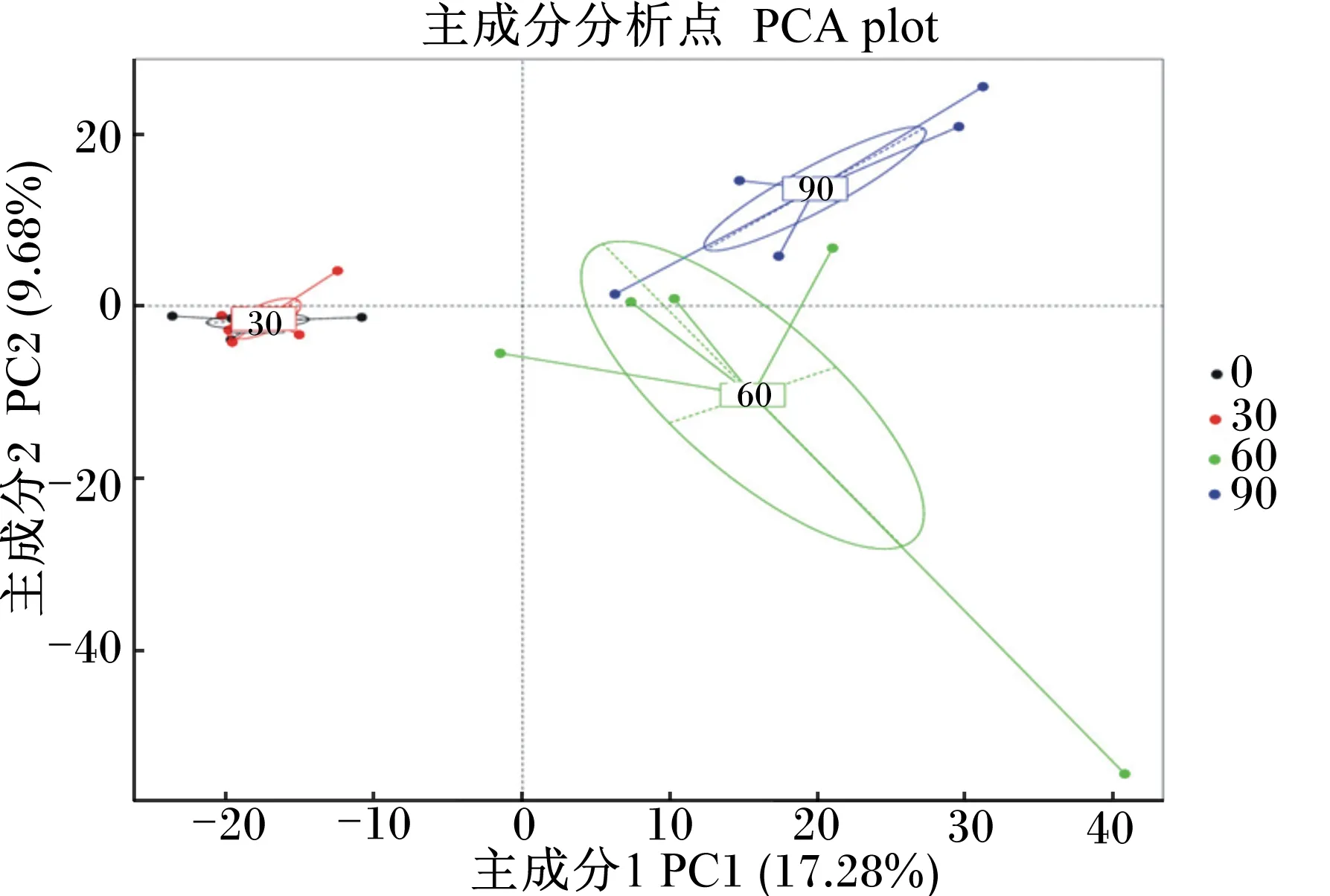
图1 主成分分析结果
2.3 马驹肠道微生物构成及分布丰度
综合分析测序样品的序列数,本试验研究了4个时间点马驹粪便中微生物群落结构,同时各时间点之间进行了比较。根据物种注释结果,选取每个样品在各分类水平上最大丰度排名前10的物种,生成物种相对丰度柱形累加图,以便直观查看各样品在不同分类水平上相对丰度较高的物种及其比例。
2.3.1 3~6月龄马驹门水平上肠道微生物丰度
在门水平上马驹肠道微生物丰度数据见表4。由表中数据可知,马驹粪便中10大优势菌门分别为厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、疣微菌门(Verrucomicrobia)、螺旋体菌门(Spirochaetes)、无壁菌门(Tenericutes)、梭杆菌门(Fusobacteria)、放线菌门(Actinobacteria)、TM7、广古菌门(Euryarchaeota),占菌群分类的98%以上,1%左右的为其他未被分类的菌门,门水平上肠道微生物丰度柱状图如图3所示。其中厚壁菌门、拟杆菌门、变形菌门及疣微菌门为优势菌门。随着月龄的增加,马驹粪便中各微生物的丰度发生了不同程度的变化。厚壁菌门的丰度在试验第0天、第30天、第60天和第90天时分别为33.19%、32.90%、73.72%和67.89%,第60天时极显著高于第0天和第30天时(P<0.01),第90天时显著高于第0天和第30天时(P<0.05)。放线菌门的丰度在试验第60天时达到1.34%,显著高于试验第0天和第30天时(P<0.05)。无壁菌门的丰度随着马驹月龄的增加而增加,在试验第90天时为3.05%,显著高于第0天和第30天时(P<0.05)。拟杆菌门的丰度在试验第0天、第30天、第60天和第90天时分别为40.97%、43.44%、0.49%和2.75%,第60天和第90天时极显著低于第0天和第30天时(P<0.01)。螺旋体菌门的丰度随着马驹月龄的增加先降低后趋于平稳,试验第0天时为3.13%,与之相比,在试验第30天、第60天和第90天时显著降低(P<0.05)。变形菌门、疣微菌门、梭杆菌门、TM7、广古菌门的丰度在整个试验期无显著性变化(P>0.05),其中变形菌门的丰度在试验第30天时降低,第60天和第90天时又有所升高,与第0天时接近。
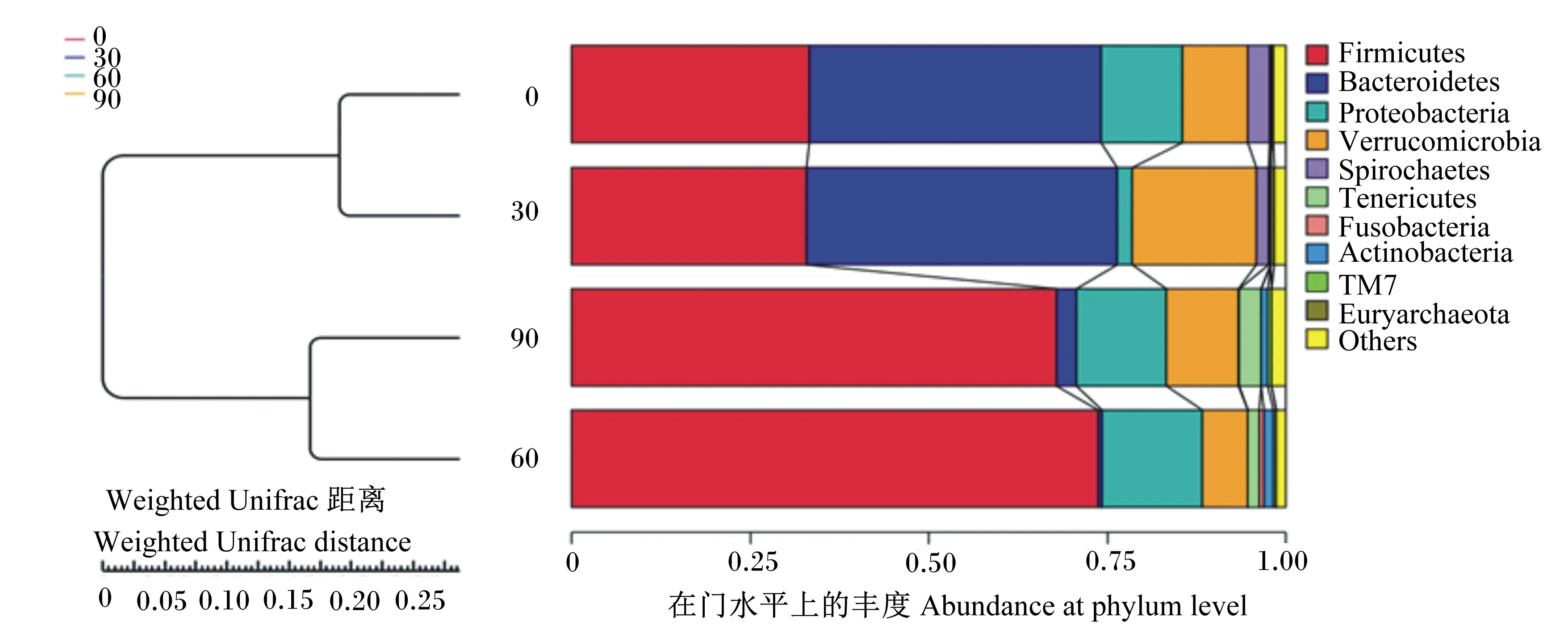
Firmicutes:厚壁菌门;Bacteroidetes:拟杆菌门;Proteobacteria:变形菌门;Verrucomicrobia:疣微菌门;Spirochaetes:螺旋体菌门; Tenericutes:无壁菌门;Fusobacteria:梭杆菌门;Actinobacteria:放线菌门;Euryarchaeota:广古菌门;Others:其他。图3同 The same as Fig.3.
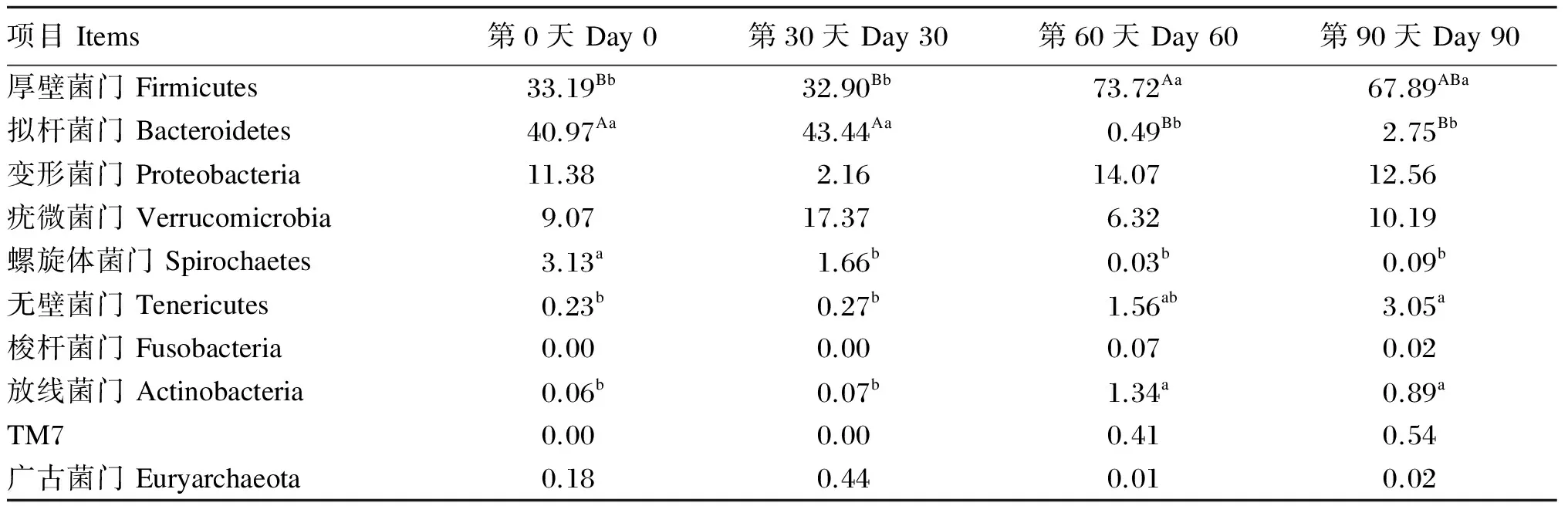
图2 基于Weighted Unifrac距离的UPGMA聚类树
同行数据肩标无字母或相同字母表示差异不显著(P>0.05),不同小写字母表示差异显著(P<0.05),不同大写字母表示差异极显著(P<0.01)。下表同。
In the same row, values with no letter or the same letter superscripts mean no significant difference (P>0.05), while with different small letter superscripts mean significant difference (P<0.05), and with different capital letter superscripts mean significant difference (P<0.01). The same as below.
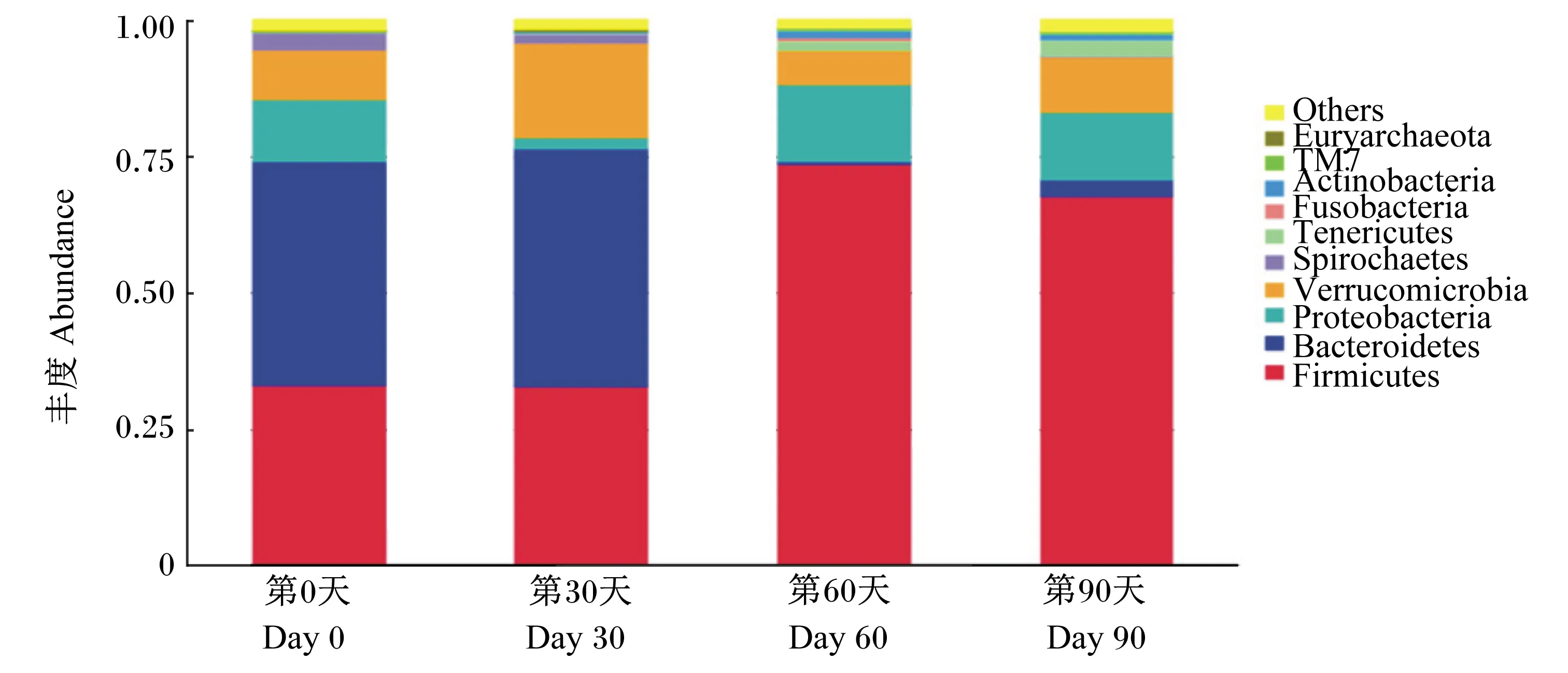
图3 3~6月龄马驹门水平上肠道微生物丰度柱状图
2.3.2 3~6月龄马驹科水平上肠道微生物丰度
在科水平上马驹肠道微生物丰度数据见表5。由表中数据可知,马驹粪便中10大优势菌科分别为芽孢杆菌科(Bacillaceae)、莫拉氏菌科(Moraxellaceae)、动球菌科(Planococcaceae)、肉杆菌科(Carnobacteriaceae)、BS11、RFP12、乳杆菌科(Lactobacillaceae)、瘤胃球菌科(Ruminococcaceae)、毛螺旋菌科(Lachnospiraceae)、紫单胞菌科(Porphyromonadaceae),科水平上肠道微生物丰度柱状图如图4所示。芽孢杆菌科的丰度在试验第60天时为22.31%,极显著高于第0天和第30天时(P<0.01),显著高于第90天时(P<0.05)。乳杆菌科的丰度在试验第90天时为5.77%,极显著高于第0天和第30天时(P<0.01)。瘤胃球菌科的丰度在试验第0天、第30天、第60天和第90天时分别为11.93%、10.19%、3.44%和7.66%,第60天时极显著低于第0天和第30天时(P<0.01),显著低于第90天时(P<0.05)。毛螺旋菌科的丰度在试验第0天、第30天、第60天和第90天时分别为6.98%、6.45%、2.96%和6.88%,第60天时显著低于第0天、第30天和第90天时(P<0.05)。紫单胞菌科的丰度在试验第0天、第30天、第60天和第90天时分别为2.58%、0.40%、0.02%和0.14%,第60天时极显著低于第0天和第30天时(P<0.01)。BS11的丰度在试验第0天、第30天、第60天和第90天时分别为8.95%、15.98%、0.06%和0.53%,第60天和第90天时显著低于第0天和第30天时(P<0.05)。莫拉氏菌科、动球菌科、肉杆菌科、RFP12的丰度在整个试验期无显著性变化(P>0.05)
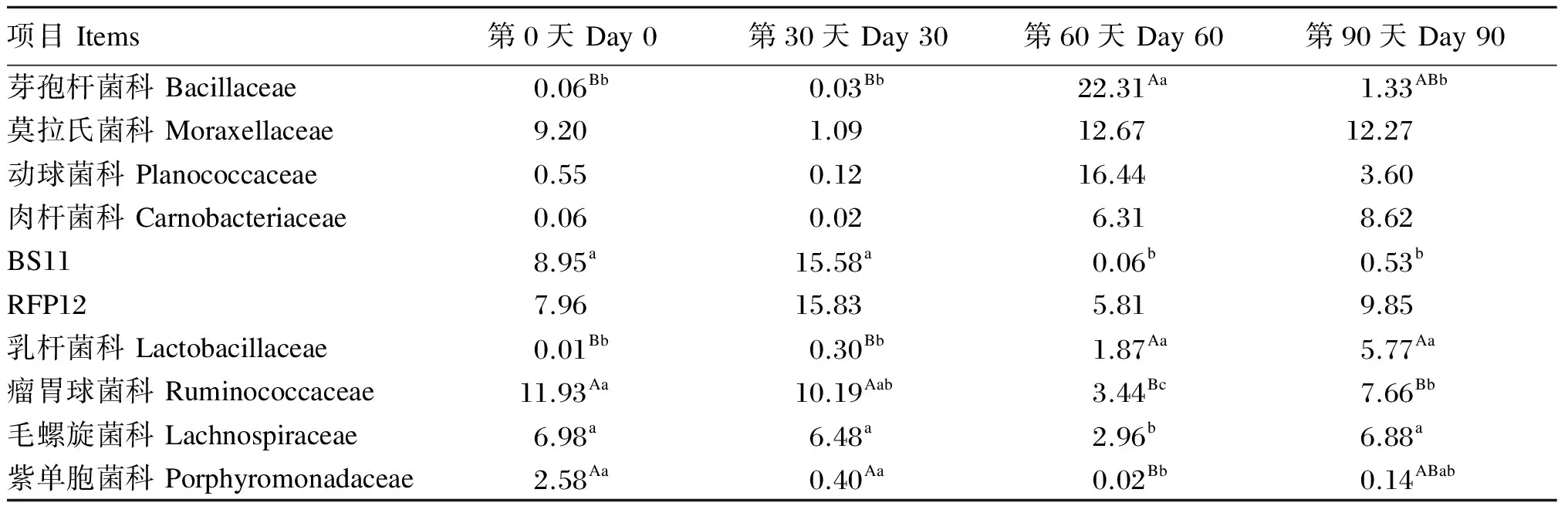
表5 3~6月龄马驹科水平上肠道微生物丰度
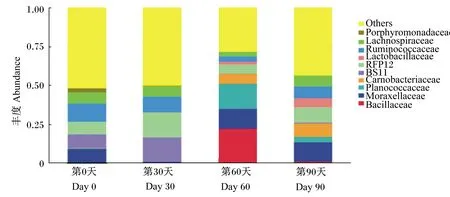
Others:其他;Porphyromonadaceae:紫单胞菌科;Lachnospiraceae:毛螺旋菌科;Ruminococcaceae:瘤胃球菌科;Lactobacillaceae:乳杆菌科;Carnobacteriaceae:肉杆菌科;Planococcaceae:动球菌科;Moraxellaceae:莫拉氏菌科;Bacillaceae:芽孢杆菌科。
图4 3~6月龄马驹科水平上肠道微生物丰度柱状图
Fig.4 The histograms of abundances of micrbiota in intestine of 3- to 6-month-old colts at family level
2.3.3 3~6月龄马驹属水平上肠道微生物丰度
在属水平上马驹肠道微生物丰度数据见表6。由表中数据可知,马驹粪便中10大优势菌属分别是不动杆菌属(Acinetobacter)、德库菌属(Desemzia)、乳杆菌属(Lactobacillus)、Ureibacillus、Paludibacter、芽孢杆菌属(Bacillus)、埃希式杆菌属(Escherichia)、肉食杆菌属(Carnobacterium)、密螺旋体属(Treponema)、艰难杆菌属(Mogibacterium),属水平上肠道微生物丰度柱状图如图5。乳杆菌属的丰度在试验第90天时为5.77%,极显著高于第0天、第30天和第60天时(P<0.01)。芽孢杆菌属的丰度在试验第60天时为4.11%,极显著高于第0天和第30天时(P<0.01)。
3 讨 论
3.1 3~6月龄马驹肠道微生物群落多样性
近年,Illumina高通量测序技术在微生物研究领域广泛使用。利用Illumina高通量测序技术能够更深入、直观、准确地通过肠道微生物群落的稳定性和多样性的变化反映动物的胃肠道健康和生长发育情况[3,6-8]。Proughbred等[9]对8匹去势纯血马的14份粪样进行测序,共获得有效序列488 213条,OTUs的平均范围为1 200~3 000个。Yatsunenk等[10]研究显示,应用高通量测序技术在人粪便中共检测出97%以上覆盖率的微生物细菌,OTUs超过2 000个。Costa等[11]在11匹马粪便中共获得有效序列数6 536 523条。本试验结果显示,通过Illumina HiSeq测序获得5匹马驹4个阶段20个粪样细菌16S rDNA有效序列数157 665条,且随着马驹月龄的增加获得有效序列数在增加,从试验第0天的23 549条增加到第90天的53 617条;平均获得1 117个OTUs,且OTUs数也随着马驹月龄的增加在增加,从试验初的1 005个增加到试验结束时的1 347个。通过α多样性分析,本试验结果显示,随着马驹月龄的增加,粪便样品的α多样性指数中ACE、Chao1、Shannon指数逐渐升高,Simpson指数则随着马驹月龄的增加逐渐降低,说明随着马驹月龄的增长、饲粮的变更,马驹肠道微生物种类越来越丰富,与Sakaitani等[4]报道的结果一致。随着马驹月龄的增加,马驹采食的固体饲草料成为肠道系统营养物质的主要来源,饲粮类型的变更是影响肠道微生物结构变化的主要因素[12]。本试验中,马驹逐渐用精料补充料及苜蓿干草替代母乳,随着母马泌乳量的减少,马驹在补喂定量精料补充料的基础上,采食的粗饲料(苜蓿干草)增加,饲粮类型逐渐改变。因此,随着马驹的生长,马驹粪便中检测出微生物的种类和数量更丰富、更多样、更复杂。该结果与Combes等[13]在家兔上的研究结果一致,即日龄对肠道菌群结构、多样性和复杂性有显著影响,其原因与饲粮的改变有直接的关系。

表6 3~6月龄马驹属水平上肠道微生物丰度
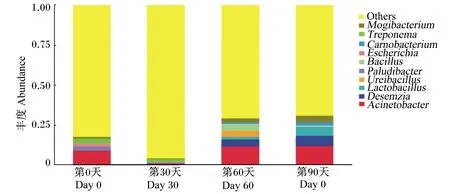
Others:其他;Mogibacterium:艰难杆菌属;Treponema:密螺旋体属;Carnobacterium:肉食杆菌属;Escherichia:埃希式杆菌属;Bacillus:芽孢杆菌属;Lactobacillus:乳杆菌属;Desemzia:德库菌属;Acinetobacter:不动杆菌属。
Others:其他;Mogibacterium:艰难杆菌属;Treponema:密螺旋体属;Carnobacterium:肉食杆菌属;Escherichia:埃希式杆菌属;Bacillus:芽孢杆菌属;Lactobacillus:乳杆菌属;Desemzia:德库菌属;Acinetobacter:不动杆菌属。
图5 3~6月龄马驹属水平上微生物丰度柱状图
Fig.5 The histograms of abundances of micrbiota in intestine of 3- to 6-month-old colts by genera level
3.2 3~6月龄马驹肠道微生物群落结构的变化
马肠道微生物定植的研究相对于反刍动物较少。马驹胎粪中没有细菌,与马驹肠道无菌结果相一致[4]。乳酸杆菌、梭状芽胞杆菌、肠杆菌在马驹出生后第3天从粪便中分离出来,12周龄的马驹肠道纤维水解菌和乳酸菌的水平和成年马相似[14]。当马驹到42日龄时,在其粪便中检测到比母马更丰富的微生物种群,且与母马的相似度达到60%。在其他一些研究中,马驹在42~80日龄与母马肠道微生物种群相似度达到50%。因此,研究马驹肠道微生物区系早期的建立对马驹肠道健康及生长发育意义重大。
许宇静等[15]采用Illumina高通量测序技术研究断奶幼兔盲肠微生物区系,发现其在门水平上主要由厚壁菌门、拟杆菌门、变形菌门和疣微菌门组成;在科水平上主要由瘤胃球菌科、毛螺旋菌科、拟杆菌科和紫单胞杆菌科组成。白秀娟等[16]利用聚合酶链式反应-变性梯度凝胶电泳(PCR-DGGE)技术分析发现,断奶仔兔肠道主要优势菌为厚壁菌门、毛螺旋菌科等。谷莉[17]采用454高通量测序技术分析发现,不同饮食结构条件下小鼠肠道厚壁菌门和拟杆菌门为主要优势菌群。本试验结果显示,采用Illumina HiSeq测序技术在马驹粪便中检测到丰度占99%以上门为厚壁菌门、拟杆菌门、变形菌门、疣微菌门、螺旋体菌门、无壁菌门、放线菌门、TM7、广古菌门,从丰度来看厚壁菌门、拟杆菌门、变形菌门、螺旋体菌门为主要的优势菌门。与以上结果对比发现,厚壁菌门和拟杆菌门是单胃大型动物和小型动物肠道主要的优势菌。本试验中,马驹在门水平上发现较多的菌群,这与种间差异有关。Dougal等[6]研究结果显示,在5匹纯血马和5匹矮种马的大肠内容物中检测到的主要优势菌为厚壁菌门、拟杆菌门、螺旋体菌门、变形菌门和放线菌门。由此说明在相同的种属间肠道微生物菌群组成更接近。
此外,在鼠、猪、马、牛等的研究中发现,厚壁菌门和拟杆菌门是其胃肠道主要的优势菌群[18-20],且厚壁菌门是摄食纤维饲料动物胃肠道促进纤维分解的主要菌群[21]。厚壁菌门能从纤维饲料中获取更多能量满足动物生长需要[22]。胃肠道菌群的丰度与消化能力有直接的关系[23-24]。厚壁菌门为马驹优势菌群,这与盲肠、结肠为马属动物主要的消化场所有关。拟杆菌门虽然随着马驹月龄的增加丰度有所降低,但仍然是马驹肠道的优势菌群。拟杆菌门是草食动物利用碳水化合物的主要菌群[25],这与马驹摄入纤维饲料有直接的关系。在单胃动物猪的研究中,饲粮中添加抗生素会减少拟杆菌门的数量[26]。本试验精料补充料中并未添加有抗生素成分的药品,但在试验前母马使用过驱虫药(主要成分为碘醚柳胺),是否因为这个原因导致马驹肠道拟杆菌门丰度降低有待于进一步的研究。
本试验中,隶属于厚壁菌门的芽孢杆菌科、乳杆菌科和瘤胃球菌科,隶属于拟杆菌门的紫单胞菌科及螺旋体菌门的毛螺旋菌科随着马驹月龄的增加呈显著性变化。研究显示,草食动物反刍或非反刍类肠道内包括毛螺旋科、纤维杆菌属等在内的细菌与其食草特性相关[27-28]。12周龄(3月龄左右)的马驹肠道纤维降解菌数量和母马相似[4]。在科水平上这些细菌丰度的变化与饲粮纤维含量的增加有直接的关系。毛螺旋菌科的细菌参与纤维素的降解[29],其主要作用是降解植物饲料细胞壁中的果胶、果糖、纤维二糖等,产生挥发性脂肪酸(乙酸、丙酸、丁酸等),为动物及肠道微生物提供能量。研究表明,饲粮变更是导致肠道微生物改变的首要原因[12]。在本试验中,随着母马泌乳量减少,马驹采食固体饲粮的量逐渐增加,故而纤维摄入量增加,这可能是促进与纤维降解相关菌种丰度增加的原因。
此外,变形菌门是动物消化道的优势菌门,其菌群庞大、种类多样,在临床研究中对胃肠道疾病诊断意义重大。很多致病菌包括大肠杆菌、沙门氏杆菌、霍乱弧菌、幽门螺杆菌都属于变形菌门的γ-变形纲[30]。本试验结果显示,变形菌门在整个试验期间丰度没有发生显著性变化,在科、属水平上排在前10的优势菌群中未发现属于变形菌门的有害菌。由此说明,随着马驹月龄的增加,厚壁菌门、拟杆菌门等有益菌丰度的增加抑制或稳定了属于变形菌门的有害菌丰度的增加,从而维持马驹肠道微生物细菌的稳定性,保证马驹肠道的健康。
4 结 论
① 本试验采用Illumina HiSeq测序技术获得获得5匹马驹4个时间点的20个粪样的有效序列数为157 665条,平均OTUs 1 117个,满足测序条件。
② 厚壁菌门、拟杆菌门、变形菌门、疣微菌门为3~6月龄伊犁马肠道主要的优势菌群。
[1] PIETZAK M.Bacterial colonization of the neonatal gut[J].Journal of Pediatric Gastroenterology and Nutrition,2004,38(4):389-391.
[2] GORBACK S L,PLAUT A G,HAHAS L,et al.Studies of intestinal microflora.Ⅱ.Microorganisms of the small intestine and their relations to oral and fecal flora[J].Gastroenterology,1967,53(6):856-867.
[3] DALY K,STEWART C S,FLINT H J,et al.Bacterial diversity within the equine large intestine as revealed by molecular analysis of cloned 16S rRNA genes[J].FEMS Microbiology Ecology,2001,38(2/3):141-151.
[4] SAKAITANI Y,YUKI N,NAKAJIMA F,et al.Colonization of intestinal microflora in newborn foals[J].Journal of Intestinal Microbiology,1999,13(1):9-14.
[5] BLASER M J,FALKOW S.What are the consequences of the disappearing human microbiota[J].Nature Reviews Microbiology,2009,7(12):887-894.
[6] DOUGAL K,DE AL FUENTE G,HARRIS P A,et al.Identification of a core bacterial community within the large intestine of the horse[J].PLoS One,2013,8(10):e77660.
[7] O’DONNELL M M,HARRIS H M B,JEFFERY I B,et al.The core faecal bacterial microbiome of Irish Thoroughbred racehorses[J].Letters in Applied Microbiology,2013,57(6):429-501.
[8] STEWART C J,MARRS E C L,MAGORRIAN S,et al.The preterm gut microbiota:changes associated with necrotizing enterocolitis and infection[J].Acta Paediatrica,2012,101(11):1121-1127.
[9] PROUGHBRED C J,HUNTER J O,DARBY A C,et al.Characterisation of the faecal metabolome and microbiome of thoroughbred racehorses[J].Equine Veterinary Journal,2015,47(5):580-586.
[10] YATSUNENK O T,REY F E,MANARY M J,et al.Human gut microbiome viewed across age and geography[J].Nature,2012,486(7402):222-227.
[11] COSTA M C,STMPFLI H R,ALLEN-VERCOE E,et al.Changes in the equine fecal microbiota associated with the use of systemic antimicrobial drugs[J].BMC Veterinary Research,2015,11:19.
[12] FONTY G,CHAUCHEYRAS-DURAND F.Lescosystèmes digestifs[M].Pairs:Tech & Doc Publishing,2007.
[13] COMBES S,MICHELLAND R J,MONTEILS V,et al.Postnatal development of the rabbit caecal microbiota composition and activity[J].FEMS Microbiology Ecology,2011,77(3):680-689.
[14] JULLIAND V,DE VAUX A,VILLARD L,et al.Preliminary studies on the bacterial flora of faeces taken from foals,from birth to twelve weeks.Effect of the oral administration of a commercial colostrum replace[J].Pferdeheilkunde,1996,12(3):209-212.
[15] 许宇静,张煜坤,沈雪梅,等.采用Illumina MiSeq测序技术分析断奶幼兔盲肠微生物群落的多样性[J].动物营养学报,2015,27(9):2793-2802.
[16] 白秀娟,刘诚刚,杜智恒,等.PCR-DGGE技术分析断奶仔兔肠道微生物菌群结构及多样性[J].东北农业大学学报,2012,43(9):64-69.
[17] 谷莉.不同鼠种的肠道菌群在不同饮食结构干预中的组成改变[D].博士学位论文.长沙:中南大学,2014.
[18] LEY R E,LOZUPONE C A,HAMADY M,et al.Worlds within worlds:evolution of the vertebrate gut microbiota[J].Nature Reviews Microbiology,2008,6(10):776-788.
[19] CALLAWAY T R,DOWD S E,EDRINGTON T S,et al.Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing[J].Journal of Animal Science,2010,88(12):3977-3983.
[20] DE MENEZES A B,LEWIS E,O’DONOVAN M,et al.Microbiome analysis of dairy cows fed pasture or total mixed ration diets[J].FEMS Microbiology Ecology,2011,78(2):256-265.
[21] BRULC J M,ANTONOPOULOS D A,MILLER M E B,et al.Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases[J].Proceedings of the National Academy of Sciences of the United States of America,2009,106(6):1948-1953.
[22] RAY A K,BAIRAGI A,SARKAR G K,et al.Optimization of fermentation conditions for cellulase production byBacillussubtilisCY5 andBacilluscirculansTP3 isolated from fish gut[J].Acta Ichthyologica et Piscatoria,2007,37(1):47-53.
[23] JIAO J Z,WANG P P,HE Z X,et al.Invitroevaluation on neutral detergent fiber and cellulose digestion by post-ruminal microorganisms in goats[J].Journal of the Science of Food and Agriculture,2014,94(9):1745-1752.
[24] LOOFT T,ALLEN H K,CANTAREL B L,et al.Bacteria,phages and pigs:the effects of in-feed antibiotics on the microbiome at different gut locations[J].The ISME Journal,2014,8(8):1566-1576.
[25] SPENCE C,WELLS W G,SMITH C J.Characterization of the primary starch utilization operon in the obligate anaerobeBacteroidesfragilis:regulation by carbon source and oxygen[J].Journal of Bacteriology,2006,188(13):4663-4672.
[26] LOOFT T,JOHNSON T A,ALLEN H K,et al.In-feed antibiotic effects on the swine intestinal microbiome[J].Proceedings of the National Academy of Sciences of the United States of America,2012,109(5):1691-1696.
[27] BIAN G R,MA L,SU Y,et al.The microbial community in the feces of the white rhinoceros (Ceratotheriumsimum) as determined by barcoded pyrosequencing analysis[J].PLoS One,2013,8(7):e70103.
[28] DALY K,PROUDMAN C J,DUNCAN S H,et al.Alterations in microbiota and fermentation products in equine large intestine in response to dietary variation and intestinal disease[J].British Journal of Nutrition,2012,107(7):989-995.
[29] SHINKAI T,UEKI T,KOBAYASHI Y.Detection and identification of rumen bacteria constituting a fibrolytic consortium dominated byFibrobactersuccinogenes[J].Animal Science Journal,2010,81(1):72-79.
[30] EVANS N J,BROWN J M,MURRAY R D,et al.Characterization of novel bovine gastrointestinal tractTreponemaisolatesand comparison with bovine digital dermatitis treponemes[J].Applied and Environmental Microbiology,2011,77(1):138-147.
*Corresponding author, professor, E-mail: yangkailun2002@aliyun.com
(责任编辑 菅景颖)
A Study on Intestinal Microbiota Diversity of 3- to 6-Month-OldYiliHorses
LI Xiaobin ZHAO Guodong LIU Zhen WU Tingting HE Zhourui DENG Haifeng YANG Kailun*
(XinjiangKeyLaboratoryofHerbivoreNutritionforMeat&MilkProduction,XingjiangAgriculturalUniversity,Urumqi830052,China)
The current study was conducted to evaluate the intestinal microbiota diversity of 3- to 6-month-oldYilihorses, to reveal the regulation of succession of intestinal micrbiota before weaning, and to provide a theoretical basis for the researches of growth and intestinal health of young horse. Five 3-month-oldYilihorses with the average body weight of (89.75±8.81) kg and the same birth day were selected to conduct a 90-day feeding trial. The feces samples were collected on days 0 (1 day before the experiment), 30, 60 and 90 of the experiment, respectively. The microbial genomic total DNA was extracted from each feces sample. Microbiota diversity of feces was evaluated using Illumina HiSeq sequencing technology. The results showed as follows: 1) twenty feces samples for 5 colts were sequenced, a total of 157 665 effective sequences and 1 117 average operational taxonomic units (OTUs) were acquired. 2) The alpha diversity indexes (ACE, Chao1, Shannon and Simpson indices) of feces microbiota showed fluctuated changes with the age increasing, but the changes were not significant among different time points (P>0.05). 3) At the phylum level, ten dominant bacteria in feces of colts were Firmicutes, Bacteroidetes, Proteobacteria, Verrucomicrobia, Spirochaetes, Fusobacteria, Tenericutes, Actinobacteria, TM7 and Euryarchaeota, and the abundances of Firmicutes, Bacteroidetes, Proteobacteria and Verrucomicrobia were higher. At the family level, ten dominant bacteria in feces of colts were Bacillaceae, Moraxellaceae, Planococcaceae, Carnobacteriaceae, BS11, RFP12, Lactobacillaceae, Ruminococcaceae, Lachnospiraceae and Porphyromonadaceae. At the genera level, ten dominant bacteria in feces of colts wereAcinetobacter,Desemzia,Lactobacillus,Ureibacillus,Paludibacter,Bacillus,Escherichia,Carnobacterium,TreponemaandMogibacterium. Base on the results, it is concluded that the intestinal microbiota of 3- to 6-month-oldYilihorses can be accurately classified using Illumina HiSeq sequencing technology. The Firmicutes, Bacteroidetes, Proteobacteria, Verrucomicrobia were the dominant bacteria in intestine of 3- to 6-month-oldYilihorses.[ChineseJournalofAnimalNutrition, 2017, 29(5):1535-1544]
Yilihorses; Illumina HiSeq sequencing technology; intestinal microbiota; diversity
10.3969/j.issn.1006-267x.2017.05.011
2016-11-11
“十二五”农村领域国家科技计划课题(2012BAD45B01)
李晓斌(1988—),男,甘肃天水人,博士研究生,研究方向为草食动物营养代谢。E-mail: 172387243@qq.com
*通信作者:杨开伦,教授,博士生导师,E-mail: yangkailun2002@aliyun.com
S852.6
A
1006-267X(2017)05-1535-10
