宏基因组学揭示瘤胃微生物多样性及功能
2017-05-12陈忠法王佳堃浙江大学奶业科学研究所杭州3002浙江万里学院生物与环境学院宁波3500
吴 鹏 陈忠法 王佳堃(.浙江大学奶业科学研究所,杭州3002;2.浙江万里学院生物与环境学院,宁波3500)
宏基因组学揭示瘤胃微生物多样性及功能
吴 鹏1陈忠法2*王佳堃1
(1.浙江大学奶业科学研究所,杭州310012;2.浙江万里学院生物与环境学院,宁波315100)
反刍动物瘤胃内栖息着庞大和复杂的微生物群体,这些微生物与宿主的消化吸收、营养代谢和免疫功能息息相关,宿主及其微生物共同组成了一个“超级生物体”。由于绝大部分瘤胃微生物不可培养,因此以厌氧培养为基础的传统研究方法存在明显的弊端。宏基因组学通过高通量的测序方法,能够全面展示微生物多样性,准确发现新的功能基因。此外,宏基因组学揭示了宿主基因和微生物组之间的互作关系。随着组学技术的不断发展,宏基因组学在瘤胃微生物组研究方面具有广阔的应用前景。
宏基因组学;瘤胃;微生物组
反刍动物全球总量已超过44亿头,是重要的食品和经济来源[1]。瘤胃容量大,温度适宜,为微生物提供了良好生长条件。瘤胃内微生物种类多,数量大,共有超过3 000种微生物,既包括细菌、原虫、真菌和古细菌[2],也有噬菌体和病毒[3]。研究表明,每毫升瘤胃液中约含1011个细菌、1010个噬菌体、109个古细菌、106个原虫和106个真菌孢子[4]。瘤胃微生物与宿主的营养、代谢和免疫息息相关[5]。瘤胃微生物利用饲粮中纤维素、半纤维素和蛋白质,产生挥发性脂肪酸(VFA),合成菌体蛋白,为宿主提供营养物质。同时,甲烷菌利用二氧化碳和氢气生成甲烷,影响全球气候变化[6]。此外,瘤胃微生物通过降解饲粮中有毒有害物质[7],产生甘露寡糖等免疫保护物质[8],可维持机体健康。瘤胃微生物调控不当,将影响动物健康。如精料采食比例过高,乳酸菌急剧增长,产生大量乳酸,导致革兰氏阴性菌死亡,释放细胞壁上的脂多糖,造成瘤胃酸中毒[9]。
宏基因组学由Handelsman等[10]于1998年提出,指对生境中所有微生物的基因组DNA进行研究,从而打破传统培养方法的局限。Lederberg[11]用“超级生物体(superorganism)”来定义宿主与共生微生物的紧密关系,反刍动物也拥有宿主本身和其共生微生物组2套遗传物质。随着人类肠道宏基因组计划[12](metagenomics of the human intestinal tract,MetaHIT)的实施和完成,人们对宿主和微生物之间的关系有了更深的认识。澳大利亚Rumen Pangenome、欧洲RuminOmics等几个研究组也已利用宏基因组学方法开展了关于反刍动物瘤胃微生物群落组成、系统发生、代谢功能的研究。如图1所示,宏基因组学起步晚,但发展迅速,目前已成为全球的研究热点。
1 宏基因组学方法操作流程
早期宏基因组学技术主要用于发掘不同生境下微生物编码特殊生物分子的基因,其一般操作流程为:首先提取环境样本中总DNA,然后将其转入Fosmid、Cosmid和细菌人工染色体等表达载体中构建宏基因组文库,最后将构建好的载体导入大肠杆菌等宿主菌中表达出活性产物。目前有基于序列、功能和底物诱导基因表达3种对宏基因组文库进行筛选的方法。Ufarté等[13]对Fosmid文库的构建方法和碳水化合物活性酶功能基因筛选方案做了较为详细的描述。Daniel[14]也介绍了目前主要文库构建和功能筛选的方法。至今,已成功将宏基因组学技术应用于土壤、瘤胃等生境中的脂酶[15]和纤维素酶[16]的发掘。
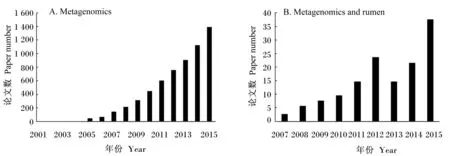
图1 PubMed中搜索关键词“Metagenomics”(A)和“Metagenomics and rumen”(B)论文数变化
随着二代测序技术的兴起,宏基因组学的内容得到不断丰富,扩增子(amplicon)测序和全基因组测序成为宏基因组研究的2个主要方向。扩增子测序可以展示微生物群落结构和丰度信息,而全基因组测序可以进一步对微生物群落功能进行分析。应用Illumina和Roche 454 FLX等测序平台对微生物宏基因组16/18S小亚基(small subunit,SSU)rRNA基因等其他系统发育标记基因进行测序,将读长(reads)序列除杂、去嵌合体后,用Mothur[17]或QIIME[18]生成可操作分类单元(operational taxonomic unit,OTU),再分别从各个OTU中挑选1条代表序列与Greengenes[19]、SILVA[20]和RDP[21]等微生物数据库比对,得到物种分类和丰度信息。Di Bella等[22]介绍了目前几种常见的高通量测序平台,并对各种测序平台的应用情况进行了说明。
此外,也有直接应用16S rRNA基因进行微生物组功能预测的报道[23]。当然,也可对样本中宏基因组DNA片段进行测序,将原始reads组装拼接形成包含更多碱基序列的重叠群(contigs)。可使用MEGAN[24]等软件将reads或contigs数据归类到对应微生物基因组上,contigs再按照一定次序排列形成scaffolds,这些scaffolds可用于构建微生物基因组草图,scaffolds间存在一定的碱基序列间隔区。reads和contigs都可用MetaGeneMark[25]或FragGeneScan[26]等软件进行基因预测,并通过与COG[27]、PFAM[28]和KEGG[29]等蛋白质、核酸及代谢相关数据库的比对注释,揭示微生物群落组成、功能和代谢特点。由于数据库种类多,操作繁琐,因此开发了MG-RAST[30]等专门用于宏基因组数据管理和分析的集成系统。Thomas等[31]针对宏基因组学研究,提供了从采样到数据分析的全套方案和建议。
2 宏基因组学揭示瘤胃微生物多样性
瘤胃微生物多样性是指对瘤胃生境中微生物组成和种群结构特点,主要包括α多样性和β多样性2个空间尺度的测定。一般用多样性指数(Chao1、ACE和Shannon指数等)作为反映丰富度和均匀度的综合指标,丰富度指微生物种类的多少,而均匀度表示各类微生物的占比情况。早期人们对瘤胃微生物的认知主要依赖于显微镜观察,因此仅局限于对一些体积较大的微生物进行形态学观察。随着研究的深入,人们很快意识到瘤胃中存在其他大量微生物,但由于培养技术所限,并不能对所有的瘤胃微生物进行分离和鉴定[4]。
得益于分子技术,特别是指纹图谱技术的发展,研究者无需对瘤胃微生物进行培养,而选择直接对微生物DNA或RNA序列进行分析,既减少了分析误差,又大大提高了对瘤胃未培养微生物的认识能力,如大型瘤胃细菌卵状奎因氏菌(Quinellaovalis)从未培养成功,但经过核糖体RNA的小亚基的基因序列分析而被确定分类地位[32]。目前用于研究微生物多样性的分子生物学技术主要包括单链构象多态性(single-strand conformation polymorphism,SSCP)、变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)、荧光原位杂交(fluorescenceinsituhybridization,FISH)、核糖体基因间隔序列分析(ribosomal intergenic sequence analysis,RISA)和可变末端-限制性片段长度多态性(terminal-restriction fragment length polymorphism,t-RFLP)[16]。这些技术在一定程度上能反映微生态群落组成结构,但分辨度仍不够高,展示的多样性信息明显低于实际值[33]。Bokulich等[34]对上述方法的优缺点进行了系统比较,并依次例举了各种方法的应用情况。
随着高通量测序技术成本的降低,宏基因组学技术的应用越来越广泛,未培养微生物也因此被大规模发现。高通量的测序技术相比于传统分子生物学方法的优势是能够直接获取碱基序列信息,而不同种类的微生物各自存在相对保守的碱基序列,亲缘关系较远,微生物保守区碱基序列的差异越大。因此,可选择这些保守区作为标记基因,宏基因组学研究中分别将序列相似性在97%、95%和90%作为在种、属和科分类水平上OTU划分的依据[2]。若无特殊说明,以下均以97%的序列相似性作为种分类水平上OTU聚类的默认阈值。
2.1 瘤胃微生物种群结构
Kim等[2]对RDP数据库中所有瘤胃来源的13 478条细菌和3 516条古菌16S rRNA基因序列进行分析。在种的分类水平上,共聚类得到5 271个细菌OTU和943个古菌OTU。其中,绝大部分细菌OTU属于厚壁菌门(2 958 OTU)、拟杆菌门(1 610 OTU)和变形菌门(226 OTU),表明瘤胃中这三大门类微生物可能占据主导地位。另外,几乎所有瘤胃来源的古菌都属于古生菌门(Euryarchaeota),甲烷短杆菌(Methanobrevibacter)是其中最主要的一类产甲烷菌。Fouts等[35]采用同样的策略,从NCBI、SILVA和RDP 3个数据库中筛选出所有瘤胃来源的细菌和古菌16S rRNA基因V1~V3区序列进行分析,共获得14 332条细菌序列和2 484条古菌序列,再分别聚类得到4 670个细菌OTU和486个古菌OTU,略低于Kim等[2]的研究结果。另外,由于RDP数据库中不包含真核生物18S序列信息,Fouts等[35]只分别从SILVA和NCBI数据库中挑选出1 027和1 803条18S序列进行真菌多样性分析,结果得到168个真菌OTU。值得注意的是,由于数据库中真菌信息并不完善,因此对于真菌比对后的分类结果和丰度信息可信度并不高[36]。也有观点认为由于真菌18S rRNA序列具有高度保守性,无法利用该序列对其进行鉴定或比较[37],因此对瘤胃真菌还有待进一步研究。
Fouts等[35]对饲喂苜蓿和干草的12头奶牛瘤胃内容物中的16/18S小亚基rRNA基因V1~V3区进行测序,分别聚类得到4 370个细菌OTU、10个古菌OTU和52个真菌OTU。与数据库中包含的所有OTU相比,各界微生物所含的OTU数均不同程度的降低,其中古菌OTU数减少最显著。在属的分类水平上,OTU数排名前5的细菌依次为普氏菌属(Prevotella)、颤杆菌属(Oscillibacter)、粪球菌属(Coprococcus)、瘤胃菌科未分类属(unclassified Ruminococcaceae)和丁酸弧菌属(Butyrivibrio),该5个属占细菌总量的40%左右。瘤胃中一共存在46个真菌属,而单个瘤胃中最多只检测到40个OTU,可见瘤胃中真菌较少。在属的分类水平上,丛赤壳属(Nectria)、Penicilliopsis、Cystofilobasidium和Delphinellan丰度较高,该4个属占真菌总数的25%以上。另外,平均每头奶牛瘤胃存在的细菌OTU数为2 122,而根据Chao1指数估算出每头牛瘤胃中包含的细菌OTU数为3 116~5 439,将近1/2的OTU序列没有检测到,表明当前测序深度(5 000条reads)并不能完全反映瘤胃细菌种群结构。而对于真菌,1 000条reads即能满足其测序要求,其覆盖度(good’s coverage)可达到98.4%以上。
Jami等[36]为探究奶牛个体间瘤胃微生物的组成差异,对饲喂相同饲粮的16头荷斯坦奶牛瘤胃内容物中微生物的16S rRNA基因V2~V3区进行扩增子测序,结果共得到4 986个OTU,平均每头牛瘤胃包含1 800个OTU。所有牛瘤胃共有的OTU数为157个(占4%),4头以上牛瘤胃所共有的OTUs数只占1/2左右。用Bray-Curtis法计算出个体间相似度为51%,个体间存在一定变异,但都包含一个由32个属组成的核心微生物组。此外,考虑到微生物间的系统发育关系,应用加权UniFrac法计算出样本间的相似度可达82%,作者指出在相同饲喂条件下虽然个体间瘤胃微生物区系在分类学上存在差异,但功能可能趋于一致。
2.2 瘤胃生境中微生物分布特点
Larue等[37]最先利用DGGE和自动核糖体基因间隔序列分析(automated ribosomal intergenic spacer analysis,ARISA)技术探究了羊瘤胃内容物固相和液相中微生物区系的组成差异。近年来,许多研究者通过高通量测序的方法,进一步证实了这种差异的存在。Mullins等[38]研究发现饲喂全混合日粮(TMR)的奶牛瘤胃内容物中,液相存在更高比例的牛链球菌(Streptococcusbovis)、栖瘤胃普氏菌(Prevotellaruminicola)和溶纤维丁酸弧菌(Butyrivibriofibrisolvens),而固相中主要以啮齿真杆菌(Eubacteriumruminantium)、产琥珀酸丝状杆菌(Fibrobactersuccinogenes)和白色瘤胃球菌(Rumincoccusalbus)为主。
McCann等[39]利用焦磷酸测序法对饲喂劣质粗饲料的肉牛瘤胃微生物展开研究,发现液相中Paludibacter和解琥珀酸菌属(Succiniclasticum)比例较固相中高,而固相中普氏菌属和螺旋体属(Treponema)丰度较高。Pitta等[40]研究了当肉牛饲粮由低蛋白质、高纤维的狗牙根(bermudagrass)向高蛋白质、高可溶性营养物的冬小麦变化时对瘤胃内容物及固相、液相微生物区系结构的影响,结果表明饲喂狗牙根草组的瘤胃内容物液相中包含的微生物种类最多,共发现149个属,而饲喂冬小麦的瘤胃内容物液相中只有118个属;普氏菌属均是存在于2种饲粮中丰度最高的微生物,且在液相分布较多,这与McCann等[39]的结果不符,可能原因在于普氏菌属存在多种代谢类型菌株,能利用不同底物生长。另外,存在于瘤胃内容物固体颗粒上的微生物可能在纤维分解方面发挥重要功能。
瘤胃上皮黏附微生物由于结构上与宿主的紧密联系,因此在功能上可能参与反刍动物瘤胃上皮VFA吸收等其他重要生理过程。Petri等[41]结合DGGE、实时荧光定量PCR(real-time quantitative PCR detecting system,qPCR)和焦磷酸测序3种技术研究了瘤胃上皮微生物在饲喂粗料和高精料时的变化规律。DGGE图谱显示饲喂粗料和混合粗料的瘤胃上皮微生物区系间相似性高于高精料组;qPCR结果表明产琥珀酸丝状杆菌的种群数量显著受到粗料的影响;焦磷酸测序结果分析得到149个OTU,厚壁菌门是存在于瘤胃上皮中的优势菌;埃氏巨球形菌(Megasphaeraelsdenii)、牛链球菌和普氏菌的qPCR定量结果与焦磷酸测序法得到的菌株丰度信息基本一致;随着饲粮的变化,瘤胃上皮微生物区系结构相较于瘤胃固相和液相更稳定[41]。
3 宏基因组学与瘤胃微生物功能
瘤胃微生物是一个极其庞大而复杂的系统,微生物间存在共生、寄生、竞争、捕食的关系。宿主健康和营养物质的消化吸收都依赖于瘤胃微生物正常功能的发挥。瘤胃微生物是一座巨大的生物资源库,蕴藏着许多有待挖掘的基因。目前,借助于宏基因组学研究手段,许多研究者对瘤胃微生物功能进行了积极探索,尝试挖掘一些重要营养生理功能密切相关的瘤胃微生物功能基因。
3.1 纤维分解
瘤胃微生物具有很强的纤维素降解能力,产生的乙酸、丙酸和丁酸等可为机体提供能量。瘤胃内的纤维降解体系非常复杂,半纤维素酶、木聚糖酶、葡糖苷酶、内切葡聚糖酶、淀粉酶、酚酸酯酶和木聚糖乙酰酯酶等一系列酶参与到这一反应过程中[42],且瘤胃微生物进化出了几种特殊纤维分解机制。纤维小体是厌氧生物存在的一种纤维素酶系,由多种纤维素酶、半纤维素酶依靠锚定、黏附机制形成的一种多酶复合体结构,通过细胞黏附蛋白附着在细菌细胞壁上,能高效彻底地降解天然纤维素材料[43]。
对高活性纤维分解基因的筛选一直是研究者致力于瘤胃微生物功能研究的主要方向。Ferrer等[44]首先在奶牛瘤胃宏基因组文库中新发现了9个内切葡聚糖酶基因。Dai等[45]为探究微生物纤维分解相关的功能基因,研究了牦牛瘤胃微生物组,试验共筛选得到223个具有纤维分解能力的细菌人工染色体(bacterial artificial chromosome,BAC)克隆,预测出10 070个开放阅读框(open reading frames,ORFs)。其中,150个ORFs属于糖苷水解酶(GH)基因,且绝大多数来自GH5、GH9和GH10家族,却缺少多数微生物纤维素酶系中重要组成成分GH48。对长度超过10 kb的水解纤维素contigs进行分析,发现其中25个来自于拟杆菌门,4个来自于厚壁菌门,且常与SusC/SusD型外膜蛋白基因连锁。Hess等[46]在瘤胃中发现27 755个碳水化合物活性基因,随机选择90个基因进行表达,57%的基因能编码具有纤维素分解活性的酶。Brulc等[47]在3头饲喂相同饲粮的肉牛瘤胃中共发现35个糖苷水解酶家族基因,然而只发现3个碳水化合物结合模块家族基因和3个锚定模块。这说明纤维降解过程中,微生物并不首先水解主链上的纤维素和半纤维素,而是先打开侧链基团,完成定植过程。
Butyrivibrioproteoclasticus是一种产丁酸的革兰氏阳性菌,可利用菊糖、果胶和木聚糖等作为发酵底物,其广泛存在于瘤胃中[33]。Kelly等[48]对ButyrivibrioproteoclasticusB316全基因组进行测序,发现其共编码3种类型的碳水化合物结合模块(CBM),分别是CBM2a、CBM6和CBM13。此外,还编码GH2、GH3、GH13等多种糖苷水解酶和糖基转移酶。
3.2 甲烷发生
Denman等[49]研究了甲烷抑制剂——溴氯甲烷(BCM-CD)对瘤胃微生物发酵的影响。BCM-CD显著增加了瘤胃氢的浓度,同时也显著提高了普氏菌和月形单胞菌等耗氢菌的丰度,从而发酵产生更多丙酸。Ross等[50]研究表明奶牛饲粮中添加葡萄酒渣和单宁与脂类物质的混合物都可减少甲烷排放,并且虽然添加剂类型不一样,但瘤胃微生物组变化特点具有相似性,2种添加剂均显著影响与甲烷生成有关的微生物,使微生物组朝着低甲烷排放的方向发展。
甲烷产量不仅与瘤胃内的甲烷菌数量相关,还受到瘤胃内其他微生物的影响。瘤胃内氢浓度和甲烷菌与产氢菌、耗氢菌的互作也是影响甲烷产量的重要因素[51]。Kittelmann等[52]研究发现瘤胃甲烷短杆菌和纤维杆菌科微生物菌群数量变化具有强正相关性,说明其在功能上存在某种联系。Ng等[53]证实瘤胃甲烷短杆菌和Butyrivibrioproteoclasticus在功能上存在一定联系。Butyrivibrioproteoclasticus可为甲烷菌甲烷发生提供甲酸、氢和二氧化碳等底物,而甲烷菌为其提供谷氨酸等营养物质。乙酸菌可以利用二氧化碳和氢生成乙酸。氢是甲烷菌生成甲烷的底物,也是其重要的能量物质[54]。乙酸菌可以与甲烷菌竞争性利用氢,从而减少甲烷的产生。但是选择合适的乙酸菌,是有效竞争氢的前提条件。Kelly等[55]通过对1株黏液细菌SA11的全基因组测序分析,发现SA11不仅可以利用二氧化碳/氢进行自养型生长,还能利用葡萄糖和甲醇进行异养型生长。但是SA11代谢类型多样,提示将其作为与甲烷菌竞争氢的候选菌可能并不合适。
为完善数据库中甲烷菌基因信息,从更深层次揭示甲烷的生成机制,目前已对包括瘤胃甲烷短杆菌M1在内的几株甲烷菌进行了全基因组测序,如表2所示。甲烷菌基因组大小不同,甲烷生成途径也各异。除二氧化碳/氢途径外,还可利用甲酸和乙酸生成甲烷。甲烷菌可能分布于瘤胃内特定的小环境,甲烷产量取决于甲烷菌对环境中营养物质的利用效率。理解甲烷菌与周围微生物的共生、互作关系,将有利于制订更加高效的甲烷减排策略。

表2 瘤胃甲烷菌全基因组测序项目
4 小结及展望
宏基因组学技术拓宽了我们对瘤胃微生态的认识,反刍动物和瘤胃微生物之间是紧密相连的整体。一个稳定、高效、平衡的微生物生态群落是瘤胃正常发挥功能的必要条件,也是决定反刍动物生产效率的重要因素。近10年来,通过宏基因组学技术对瘤胃微生态的研究,我们对反刍动物的营养机制有了大致地了解,尤其对粗饲料的消化。但相对而言,反刍动物口腔和肠道微生态的研究较少,也许这些部位微生物与瘤胃微生物之间可能存在一定的联系。
宏基因组学研究的核心是通过与现有数据库比对,从而了解系统发育和群落结构和功能信息。因此,需从完善数据库、增加测序读长、加强数据分析平台的构建等方面进一步提高宏基因组学的应用价值。但也不能彻底抛弃传统微生物研究的分离培养方法,分离培养技术的改进将大大增加瘤胃可培养微生物的种类,对纯菌生理特性和基因组信息的研究有利于对高通量测序技术组学数据的理解和解释微生物现象。Hungate 1000计划(www.hungate1000.org.nz)和FibeRumBa(http://www.jcvi.org/rumenomics)将帮助我们认识更多未培养微生物的功能及遗传机制,从而完善数据库信息。
当然,宏基因组学分析也存在一定弊端,如DNA序列只能反映微生物的代谢潜能,而不能实时反映基因的转录和表达情况,因此未来宏基因组学将联合宏转录组学、宏蛋白质组学等多组学技术,在DNA、RNA和蛋白质3个层面上揭示微生物群落的结构、系统发生、代谢功能、调控规律等。相信这些技术将会在多种生态领域得到快速的发展和应用,可以使人们从系统角度全面认识微生物群落与其功能,利用其规律挖掘新的微生物菌种及其酶资源等。随着测序成本的降低和准确性的提高,宏基因组学技术将会像PCR技术一样,成为未来微生物研究的主流方法。
[1] Food and Agriculture Organization of the United Nations.FAOSTAT[EB/OL].[2016-10-01].http://faostat3.fao.org/faostat-gateway/go/to/download/Q/QC/S.
[2] KIM M,MORRISON M,YU Z T.Status of the phylogenetic diversity census of ruminal microbiomes[J].FEMS Microbiology Ecology,2011,76(1):49-63.
[3] ROSS E M,PETROVSKI S,MOATE P J,et al.Metagenomics of rumen bacteriophage from thirteen lactating dairy cattle[J].BMC Microbiology,2013,13(1):242.
[4] KLIEVE A V,SWAIN R A.Estimation of ruminal bacteriophage numbers by pulsed-field gel electrophoresis and laser densitometry[J].Applied and Environmental Microbiology,1993,59(7):2299-2303.
[6] KUMAR S,CHOUDHURY P K,CARRO M D,et al.New aspects and strategies for methane mitigation from ruminants[J].Applied Microbiology and Biotechnology,2014,98(1):31-44.
[7] JONES R J.Does ruminal metabolism of mimosine explain the absence of leucaena toxicity in Hawaii?[J].Australian Veterinary Journal,1981,57(1):55-56.
[8] 戈婷婷.不同组合的功能性寡糖对锦江黄牛瘤胃体外发酵的影响[D].硕士学位论文.南昌:江西农业大学,2011.
[9] MALEKKHAHI M,TAHMASBI A M,NASERIAN A A,et al.Effects of supplementation of active dried yeast and malate during sub-acute ruminal acidosis on rumen fermentation,microbial population,selected blood metabolites,and milk production in dairy cows[J].Animal Feed Science and Technology,2016,213:29-43.
[10] HANDELSMAN J,RONDON M R,BRADY S F,et al.Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products[J].Chemistry & Biology,1998,5(10):R245-R249.
[11] LEDERBERG J.Infectious history[J].Science,2000,288(5464):287-293.
[12] EHRLICH S D,CONSORTIUM T M.MetaHIT:the European union project on metagenomics of the human intestinal tract[M]//NELSON K E.Metagenomics of the human body.New York:Springer,2011.
[13] UFARTÉ L,BOZONNET S,LAVILLE E,et al.Functional metagenomics:construction and high-throughput screening of fosmid libraries for discovery of novel carbohydrate-active enzymes[M]//MARTIN F,UROZ S.Microbial environmental genomics (MEG).New York:Springer,2016,1399:257.
[14] DANIEL R.The metagenomics of soil[J].Nature Reviews Microbiology,2005,3(6):470-478.
[15] DE SANTI C,ALTERMARK B,PIERECHOD M M,et al.Characterization of a cold-active and salt tolerant esterase identified by functional screening of Arctic metagenomic libraries[J].BMC Biochemistry,2016,17(1):1.
[16] PANDEY S,GULATI S,GOYAL E,et al.Construction and screening of metagenomic library derived from soil for β-1,4-endoglucanase gene[J].Biocatalysis and Agricultural Biotechnology,2016,5:186-192.
[17] SCHLOSS P D,WESTCOTT S L,RYABIN T,et al.Introducing mothur:open-source,platform-independent,community-supported software for describing and comparing microbial communities[J].Applied and Environmental Microbiology,2009,75(23):7537-7541.
[18] CAPORASO J G,KUCZYNSKI J,STOMBAUGH J,et al.QIIME allows analysis of high-throughput community sequencing data[J].Nature Methods,2010,7(5):335-336.
[19] DESANTIS T Z,HUGENHOLTZ P,LARSEN N,et al.Greengenes,a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J].Applied and Environmental Microbiology,2006,72(7):5069-5072.
[20] PRUESSE E,QUAST C,KNITTEL K,et al.SILVA:a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB[J].Nucleic Acids Research,2007,35(21):7188-7196.
[21] COLE J R,CHAI B,MARSH T L,et al.The Ribosomal Database Project (RDP-Ⅱ):previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy[J].Nucleic Acids Research,2003,31(1):442-443.
[22] DI BELLA J M,BAO Y G,GLOOR G B,et al.High throughput sequencing methods and analysis for microbiome research[J].Journal of Microbiological Methods,2013,95(3):401-414.
[23] ABHAUER K P,WEMHEUER B,DANIEL R,et al.Tax4Fun:predicting functional profiles from metagenomic 16S rRNA data[J].Bioinformatics,2015,31(17):2882-2884.
[24] HUSON D H,AUCH A F,QI J,et al.MEGAN analysis of metagenomic data[J].Genome Research,2007,17(3):377-386.
[25] TANG S,BORODOVSKY M.Ab initio gene identification in metagenomic sequences[M]//NELSON K E.Encyclopedia of metagenomics.New York:Springer,2014.
[26] RHO M,TANG H X,YE Y Z.FragGeneScan:predicting genes in short and error-prone reads[J].Nucleic Acids Research,2010,38(20):e191.
[27] TATUSOV R L,FEDOROVA N D,JACKSON J D,et al.The COG database:an updated version includes eukaryotes[J].BMC Bioinformatics,2003,4(1):41.
[28] FINN R D,ALEX B,JODY C.PFAM:the protein families database[J].Nucleic Acids Research,2014,42(D1):D222-D230.
[29] KANEHISA M,GOTO S,KAWASHIMA S,et al.The KEGG resource for deciphering the genome[J].Nucleic Acids Research,2004,32(Suppl.1):277D-280D.
[30] GLASS E M,WILKENING J,WILKE A,et al.Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes[J].Cold Spring Harbor Protocols,2010,doi:10.1101/pdb.prot5368.
[31] THOMAS T,GILBERT J,MEYER F.Metagenomics—a guide from sampling to data analysis[J].Microbial Informatics and Experimentation,2012,2(1):3.
[32] KRUMHOLZ L R,BRYANT M P,BRULLA W J,et al.Proposal ofQuinellaovalisgen. nov.,sp. nov.,based on phylogenetic analysis[J].International Journal of Systematic and Evolutionary Microbiology,1993,43(2):293-296.
[33] PAILLARD D,MCKAIN N,RINCON M T,et al.Quantification of ruminalClostridiumproteoclasticumby real-time PCR using a molecular beacon approach[J].Journal of Applied Microbiology,2007,103(4):1251-1261.
[34] BOKULICH N A,MILLS D A.Next-generation approaches to the microbial ecology of food fermentations[J].BMB Reports,2012,45(7):377-389.
[35] FOUTS D E,SZPAKOWSKI S,PURUSHE J,et al.Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen[J].PLoS One,2012,7(11):e48289.
[36] JAMI E,ISRAEL A,KOTSER A,et al.Exploring the bovine rumen bacterial community from birth to adulthood[J].The ISME Journal,2013,7(6):1069-1079.
[37] LARUE R,YU Z T,PARISI V A,et al.Novel microbial diversity adherent to plant biomass in the herbivore gastrointestinal tract,as revealed by ribosomal intergenic spacer analysis andrrsgene sequencing[J].Environmental Microbiology,2005,7(4):530-543.
[38] MULLINS C R,MAMEDOVA L K,CARPENTER A J,et al.Analysis of rumen microbial populations in lactating dairy cattle fed diets varying in carbohydrate profiles andSaccharomycescerevisiaefermentation product[J].Journal of Dairy Science,2013,96(9):5872-5881.
[39] MCCANN J C,DREWERY M L,SAWYER J E,et al.Effect of postextraction algal residue supplementation on the ruminal microbiome of steers consuming low-quality forage[J].Journal of Animal Science,2014,92(11):5063-5075.
[40] PITTA D W,PINCHAK W E,DOWD S E,et al.Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets[J].Microbial Ecology,2010,59(3):511-522.
[41] PETRI R M,SCHWAIGER T,PENNER G B,et al.Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis[J].Applied and Environmental Microbiology,2013,79(12):3744-3755.
[42] XUE G P,GOBIUS K S,ORPIN C G.A novel polysaccharide hydrolase cDNA (celD) fromNeocallimastixpatriciarumencoding three multi-functional catalytic domains with high endoglucanase,cellobiohydrolase and xylanase activities[J].Microbiology,1992,138(11):2397-2403.
[43] HAMMEL M,FIEROBE H P,CZJZEK M,et al.Structural basis of cellulosome efficiency explored by small angle X-ray scattering[J].Journal of Biological Chemistry,2005,280(46):38562-38568.
[44] FERRER M,GOLYSHINA O V,CHERNIKOVA T N,et al.Novel hydrolase diversity retrieved from a metagenome library of bovine rumen microflora[J].Environmental Microbiology,2005,7(12):1996-2010.
[45] DAI X,ZHU Y X,LUO Y F,et al.Metagenomic insights into the fibrolytic microbiome in yak rumen[J].PLoS One,2012,7(7):e40430.
[46] HESS M,SCZYRBA A,EGAN R,et al.Metagenomic discovery of biomass-degrading genes and genomes from cow rumen[J].Science,2011,331(6016):463-467.
[47] BRULC J M,ANTONOPOULOS D A,MILLER M E B,et al.Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases[J].Proceedings of the National Academy of Sciences of the United States of America,2009,106(6):1948-1953.
[48] KELLY W J,LEAHY S C,ALTERMANN E,et al.The glycobiome of the rumen bacteriumButyrivibrioproteoclasticusB316Thighlights adaptation to a polysaccharide-rich environment[J].PLoS One,2010,5(8):e11942.
[49] DENMAN S E,FERNANDEZ G M,SHINKAI T,et al.Metagenomic analysis of the rumen microbial community following inhibition of methane formation by a halogenated methane analog[J].Frontiers in Microbiology,2015,6:1087.
[50] ROSS E M,MOATE P J,MARETT L,et al.Investigating the effect of two methane-mitigating diets on the rumen microbiome using massively parallel sequencing[J].Journal of Dairy Science,2013,96(9):6030-6046.
[51] MORGAVI D P,FORANO E,MARTIN C,et al.Microbial ecosystem and methanogenesis in ruminants[J].Animal,2010,4(7):1024-1036.
[52] KITTELMANN S,SEEDORF H,WALTERS W A,et al.Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial,archaeal and eukaryotic microorganisms in rumen microbial communities[J].PLoS One,2013,8(2):e47879.
[53] NG F,KITTELMANN S,PATCHETT M L,et al.An adhesin from hydrogen-utilizing rumen methanogenMethanobrevibacterruminantiumM1 binds a broad range of hydrogen-producing microorganisms[J].Environmental Microbiology,2016,18(9):3010-3021.
[54] WEIMER P J.Redundancy,resilience,and host specificity of the ruminal microbiota:implications for engineering improved ruminal fermentations[J].Frontiers in Microbiology,2015,6:296.
[55] KELLY W J,HENDERSON G,PACHECO D M,et al.The complete genome sequence ofEubacteriumlimosumSA11,a metabolically versatile rumen acetogen[J].Standards in Genomic Sciences,2016,11(1):26.
*Corresponding author, associate professor, E-mail: 572680715@qq.com
(责任编辑 菅景颖)
Metagenomics Reveals Rumen Microbial Diversity and Functions
WU Peng1CHEN Zhongfa2*WANG Jiakun1
(1.InstituteofDairyScience,ZhejiangUniversity,Hangzhou310012,China; 2.DepartmentofBioscience,ZhejiangWanliUniversity,Ningbo315100,China)
There are a large number of microorganisms that inhabit the rumen of ruminant animals, which are closely related to host digestion, absorption and immunization. The host and symbiotic microorganisms together constitute a “superorganisms”. Because most of microorganisms in the rumen cannot be cultivated, traditional culture-dependent methods have obvious drawbacks. Metagenomics is a high throughput method that characterizes the rumen microbial community structure and identifies novel functional genes. Moreover, the interactions between microbiome and host genetics are also determined through the use of metagenomics. With advances in omic technologies, metagenomics can be a powerful tool in studying the rumen microbiome with encouraging prospects.[ChineseJournalofAnimalNutrition, 2017, 29(5):1506-1514]
metagenomics; rumen; microbiome
10.3969/j.issn.1006-267x.2017.05.008
2016-10-09
国家自然科学基金(31372337)
吴 鹏(1992—),男,湖南邵阳人,硕士研究生,从事反刍动物营养研究。E-mail: wp357@foxmail.com
*通信作者:陈忠法,副教授,硕士生导师,E-mail: 572680715@qq.com
Q78;S852.65
A
1006-267X(2017)05-1506-09
