苯和甲醇在H-ZSM-5催化剂上甲基化的反应机理
2017-05-12李玲玲张作良李晓亮聂小娃宋春山郭新闻
李玲玲 陈 韧 戴 戬 孙 野 张作良 李晓亮聂小娃 宋春山 郭新闻
苯和甲醇在H-ZSM-5催化剂上甲基化的反应机理
李玲玲1陈 韧1戴 戬1孙 野1张作良1李晓亮1聂小娃2,*宋春山2,3郭新闻2,*
(1辽宁科技学院冶金工程学院,辽宁本溪117004;2大连理工大学化工学院,精细化工国家重点实验室,PSU-DUT联合能源研究中心,辽宁大连116024;3宾夕法尼亚州立大学能源与矿物工程系能源研究所,PSU-DUT联合能源研究中心,大学城16802,宾夕法尼亚,美国)
采用“our own-N-layered integrated molecular orbital+molecular mechanics”(ONIOM)和密度泛函理论(DFT)结合的方法,在5T、12T、104T9和104T12H-ZSM-5模型中研究了苯和甲醇甲基化的分步和协同机理。描述了中间体物种和过渡态的结构。考察了H-ZSM-5催化剂Brønsted(B)酸强度对苯和甲醇甲基化反应机理的影响。反应活化能结果表明,在B酸强度更强的H-ZSM-5催化剂上,苯和甲醇甲基化反应更容易发生,反应活化能更低。随着B酸强度增强,分步机理的反应活化能比协同机理的反应活化能降低的更多。B酸强度增强对分步机理更有利。当分步机理成为主导反应路径时,分步机理中甲醇脱水步骤生成的甲氧基中间体进一步生成大体积烃类的副反应会导致H-ZSM-5催化剂因积炭而失活。合理调变H-ZSM-5催化剂的酸强度对提高催化剂的催化活性和稳定性有重要意义。
密度泛函理论;ONIOM;苯甲基化;甲醇;H-ZSM-5
1 引言
苯主要来源于煤化工中煤焦油工艺以及石油工业生产乙烯的副产物,甲醇主要来源于煤化工和天然气合成工业。工业生产中苯和甲醇产能大量过剩,导致能源和原料浪费,对苯和甲醇再利用有利于资源优化配置。苯和甲醇通过一次甲基化反应可以合成甲苯,甲苯进一步甲基化可以制备对二甲苯,对二甲苯是石油化工生产的重要原料之一,其进一步氧化和聚合可以生产聚酯材料、药物、油墨和香料等,因此苯和甲醇烷基化合成甲苯和二甲苯的反应逐渐引起了人们的关注。
ZSM-5分子筛具有特殊孔道结构及可调变的酸性中心,其作为催化剂在苯和甲醇甲基化反应中表现出良好的催化性能1-6。ZSM-5分子筛的酸强度是影响催化剂催化活性、稳定性和选择性的重要因素。ZSM-5催化苯和甲醇甲基化反应可得到甲苯、二甲苯、乙苯、甲乙苯和丙苯等产物,甲苯和二甲苯为目的产物。由于反应温度为460°C及以上时,生成甲乙苯和丙苯的反应不能自发进行,生成乙苯的反应可以自发进行,但反应程度没有芳烃甲基化反应程度高,因此乙苯为主要的副产物7。随着ZSM-5分子筛的硅铝比增加,分子筛的酸强度增加,总酸量降低,分子筛对苯甲基化反应的催化活性随之提高,苯和甲醇的转化率增加。Hu等8在ZSM-5分子筛催化苯和甲醇甲基化反应的研究中发现,增加分子筛硅铝比可以抑制乙苯生成,当ZSM-5的Si/Al为1800时,乙苯的选择性可控制在1%以下,目的产物的选择性增加。虽然增加ZSM-5分子筛硅铝比可以提高催化剂催化活性和选择性,但是分子筛更容易失活,稳定性下降。袁苹等4采用拉曼光谱和热重等方法针对ZSM-5分子筛催化苯与甲醇烷基化反应过程中积炭物种和失活机理进行了研究,发现催化剂失活主要是由反应过程中生成的大分子稠环芳烃堵塞了分子筛孔道并覆盖活性位点造成的。调变分子筛催化剂的酸中心性质是提高催化剂催化活性和稳定性及产物选择性的重要途径。
前人对分子筛催化苯及其衍生物与甲醇甲基化的反应机理进行了一系列探索,涉及了反应热力学计算、反应路径、反应能量和反应速率等科学问题9-14。目前,针对苯和甲醇甲基化反应路径和能量的理论计算研究通常在Brønsted(B)酸强度固定的分子筛模型中进行,但是不同分子筛模型活性位上B酸强度不同,苯和甲醇甲基化反应机理可能不同。至今为止,ZSM-5分子筛B酸强度对苯和甲醇甲基化反应机理的影响仍未被探究。在苯和甲醇甲基化反应中,主要副反应是生成乙苯的反应。由于甲醇自身脱水反应生成乙烯丙烯反应的吉布斯自由能小于零,甲醇可以自发反应生成乙烯,乙烯与苯进一步乙基化生成乙苯副产物7。Hansen等14在5T和17T ZSM-5分子筛簇模型中计算了苯和乙烯在B酸活性中心上乙基化生成乙苯反应的分步机理和协同机理,17T模型中分步机理和协同机理的反应活化能比5T模型中对应反应机理的反应活化能高18.0和5.0 kJ·mol-1。反应活化能计算结果表明,分子筛硅铝比增加,由于苯乙基化反应活化能增加,乙苯的生成反应被抑制。由于ZSM-5分子筛硅铝比增加,B酸强度增加会抑制乙苯副产物的生成,弄清ZSM-5分子筛活性位上B酸强度与苯和甲醇甲基化生成甲苯反应路径以及催化剂的催化活性和稳定性之间的内在联系,可以深入解释实验现象,为ZSM-5分子筛催化剂的优化设计提供理论依据。因此,本文将采用“our own-N-layered integrated molecular orbital+molecular mechanics”(ONIOM)和密度泛函理论(DFT)相结合的方法,计算H-ZSM-5催化苯和甲醇甲基化的反应机理。
2 计算模型和方法
2.1 计算模型
ZSM-5分子筛结构中一共有12种T原子,T4原子和T10原子位于直孔道的十元环上,T8原子和T11原子位于正弦形孔道的十元环上,其余8个T原子位于直孔道和正弦形孔道的交叉处15。铝原子落在位于直孔道和正弦形孔道交叉处的T6,T9和T12处的可能性较大15-18。
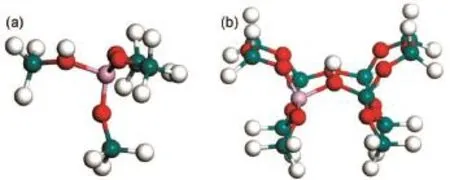
图1 两种H-ZSM-5分子筛模型Fig.1 Two models of the H-ZSM-5 molecular sieve
从MFI分子筛晶格结构中,截取图1(a)和图1 (b)所示的5T和12T分子筛计算模型19-21。在5T和12T模型中,用一个铝原子代替T12位的硅原子,同时引入一个氢原子作为B酸中和由原子替代产生的负电荷,催化剂的活性位由此产生22-24。5T和12T H-ZSM-5分子筛计算模型的硅铝比不同,活性位上B酸强度不同。
截取包含ZSM-5直孔道和正弦形孔道结构的104T H-ZSM-5分子筛模型计算苯和甲醇在不同B酸活性位上甲基化反应机理。对于分子筛孔内催化体系来说,反应机理的相关能量不仅取决于B酸性质,还与分子筛骨架结构的空间位阻限制作用有关。B酸活性位周围的分子筛孔道骨架结构环境不同,反应物和过渡态结构受到分子筛孔道骨架结构的空间位阻限制作用不同,不同的空间限制作用对反应机理相关能量的计算结果会产生较大的影响。因此,为了考察B酸强度对反应机理的影响,选取周围分子筛孔道骨架结构环境相似的T9和T12位作为铝原子的取代位。T9和T12位的硅原子分别被一个铝原子替代,对应地构建出104T9和104T12H-ZSM-5分子筛计算模型,分别如图2(c)和图2(d)所示。104T9模型和104T12模型中均引入一个氢原子(B酸)来平衡体系电荷。模型终端Si―O键被截断,用氢原子将硅原子饱和。为了防止优化时模型发生不合理变形,沿着分子筛骨架结构的方向固定Si―H键,键长为0.147 nm。由于104T9和104T12H-ZSM-5模型中铝原子的落位不同,活性位上B酸强度不同。
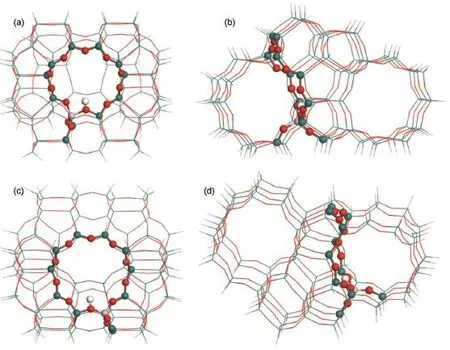
图2 包含H-ZSM-5内部结构的104T ONIOM2模型Fig.2 104T ONIOM2 model involving the internal H-ZSM-5 structure
2.2 计算方法
采用ONIOM2方法25-27计算104T(104T9和104T12)H-ZSM-5模型中平衡态和过渡态物种的几何构型。此方法将104T模型分为高层和低层两部分,将活性中心所在的10元环,铝原子相邻的两个硅原子以及物种分子作为高层,在B3LYP/6-31G (d,p)水平上计算。模型中剩余的部分(92T)作为低层,用UFF分子力场计算28。12T高层区域中的5T [(≡(SiO)3Al(OH)Si≡]和反应物分子在结构优化过程中完全松弛,其余部分被固定在晶格方向。在结构优化过程中,计算了每个驻点的频率,保证平衡态物种在反应方向上最稳定,过渡态只有一个虚频,并沿着反应方向振动。Van der Mynsbrugge等13提出,对于H-ZSM-5催化芳烃甲基化反应,采用quasi-IRC方法,通过沿着过渡态的虚频的振动方向微微扰动结构,对微扰结构进行优化,获得连接过渡态的反应物和产物结构。虽然ONIOM2方法能够很好的描述吸附物种的结构,但是在高层和低层的连接处会引入较大的计算误差,ONIOM2方法会过高地估算吸附能量,因此通常在ONIOM2方法优化好的结构基础上采用DFT方法计算单点能19-21,29,30。ωB97X-D泛函能够很好地描述主族热化学、动力学和非共价相互作用,并且能很好的描述芳烃和分子筛骨架的相互作用13,31-33。将ONIOM2方法优化好的几何结构再采用ωB97X-D泛函在6-31G(d,p)水平上计算单点能,并考虑零点能校正。
采用B3LYP/6-31G(d,p)方法对5T或12T模型、反应物、过渡态和产物进行结构优化,用ωB97X-D/6-31G(d,p)方法对优化好的平衡态结构计算单点能,并且校正零点能。所有的计算采用Gaussion 0934软件。
3 结果与讨论
3.1 5T和12T模型中苯和甲醇甲基化反应
为了比较5T和12T两种模型B酸酸性的强弱,计算了质子亲核势(PA)及NH3在活性位上的吸附能35。5T模型的PA比12T模型的PA低46.0 kJ· mol-1。PA越负,说明分子筛碱性位和B酸质子之间的相互作用越强,酸性越弱。NH3在12T模型活性位上的吸附能比在5T模型活性位上的吸附能低20.1 kJ·mol-1。吸附能越低,说明氨气吸附越稳定,B酸和氨气之间的相互作用越强,酸性越强。从PA和NH3吸附能计算结果可以看出,12T HZSM-5模型的B酸酸性更强,这与前人得到的酸强度随着硅铝比增加而增强的结论一致1-3,35,36。
H-ZSM-5分子筛催化苯和甲醇烷基化有两种可能的反应路径9-13,37。分步机理认为:首先甲醇分子在分子筛活性位上吸附。B酸质子进攻甲醇的羟基氧原子导致碳―氧键断裂,生成的甲基基团和分子筛骨架氧原子形成甲氧基中间体产物,同时释放一分子水。水分子在甲基化反应之前脱附。随后苯分子在甲氧基中间体附近吸附。甲氧基的甲基基团和吸附的苯分子发生甲基化反应,最终生成甲苯。协同机理认为:首先苯和甲醇在分子筛活性位上共吸附。接着B酸质子进攻甲醇的羟基氧原子,甲醇分子解离成一个羟基和一个甲基基团,羟基和B酸质子之间形成新的化学键,生成一分子水。与此同时,甲基基团变成sp2杂化的碳正离子结构,进攻苯分子,发生甲基化反应,生成最终的甲苯和水。协同机理中不生成甲氧基中间体产物,水分子伴随整个甲基化过程。
在5T模型中,图3(a)为苯和甲醇沿着分步机理进行甲基化反应的能线图,图中给出了甲基化步骤的过渡态结构。在分步机理中,吸附的甲醇克服207.4 kJ·mol-1能垒脱水生成甲氧基中间体和一分子水,反应吸热55.6 kJ·mol-1。随后,水分子从活性中心脱附。接着,一分子苯扩散到甲氧基中间体周围与其形成共吸附络合物结构。经过甲基化过渡态,甲氧基中间体和苯甲基化生成甲苯产物,甲基化步骤的反应活化能为162.6 kJ· mol-1。以吸附的甲醇的能量为参考点,苯和甲醇沿着分步机理发生甲基化反应的总反应活化能为236.7 kJ·mol-1。图3(b)包括了苯和甲醇沿着协同机理进行甲基化反应的能线图和过渡态结构。在协同机理中,甲醇的C―O键和苯的C―H键断裂,甲醇和苯分子之间形成C―C键。在过渡态结构中,甲醇C―O键断裂生成的甲基基团变为sp2杂化的甲基碳正离子结构,甲基碳正离子被水分子和苯环的π电子共同稳定。以苯和甲醇形成的共吸附结构的能量为参考点,苯和甲醇甲基化协同机理的总反应活化能为200.7 kJ·mol-1。比较两种反应机理的总反应活化能可以得出,5T模型中协同机理为苯和甲醇甲基化反应的主导反应路径。
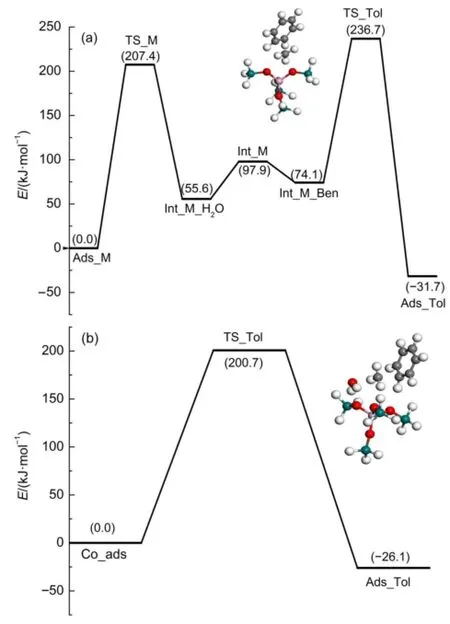
图3 5T模型中苯和甲醇甲基化反应能线图以及甲基化过渡态结构Fig.3 Reaction energy profile and methylation transition state structures for benzene methylation with methanol on the 5T model
在12T模型中,苯和甲醇沿着分步机理和协同机理进行甲基化反应的能线图分别如图4(a)和图4 (b)所示,甲基化步骤的过渡态结构在对应的能线图中给出。在分步机理中,甲醇在分子筛B酸活性位上克服157.5 kJ·mol-1能垒脱水,生成甲氧基中间体和一分子水,反应吸热26.2 kJ·mol-1。生成的水分子在甲氧基的附近吸附,并且在甲基化反应前脱附。随后,一分子苯和甲氧基以σ-π相互作用形成甲基化步骤的反应物结构。苯和甲醇分步甲基化反应的总反应活化能为184.5 kJ·mol-1。在协同机理中,甲基化始于苯和甲醇形成共吸附反应物,分子筛的B酸质子进攻甲醇的C―O键,诱导甲醇脱水和甲基化反应发生。在甲基化过渡态结构中,甲醇的C―O键完全断裂,产物水已经生
图4 12T模型中苯和甲醇甲基化反应能线图以及甲基化过渡态结构
Fig.4 Reaction energy profile and methylation transition state structures for benzene methylation with methanol on the 12T model成,甲基碳正离子和苯环沿着生成新的C―C键的方向相互作用。协同机理的总反应活化能为189.5 kJ·mol-1,比分步机理的总反应活化能高5.0 kJ· mol-1。可以得出,在12T模型中,分步机理和协同机理的反应活化能相差不大,两条反应路径为竞争关系。
比较5T模型和12T模型中苯和甲醇甲基化的分步机理和协同机理的反应活化能结果可得,12T模型中分步机理和协同机理的反应活化能分别比5T模型中对应反应机理的反应活化能低52.5和11.2 kJ·mol-1,证明随着B酸酸性增强,H-ZSM-5分子筛对苯甲基化反应的催化活性提高,这与前人对苯和甲醇甲基化的实验研究观察到的实验现象一致,随着ZSM-5分子筛硅铝比增加,苯和甲醇的转化率都增加1,2。在12T模型中,分步机理的反应活化能比协同机理的反应活化能低5.0 kJ· mol-1。在5T模型中,协同机理的反应活化能比分步机理的反应活化能低36.0 kJ·mol-1。Vos等12在4T H-ZSM-5(AlSi3O4H10)模型中计算的苯甲基化协同机理的反应活化能比分步机理的反应活化能低59.97 kJ·mol-1。4T模型和5T模型中协同机理为主导反应路径,12T模型中分步机理已经成为协同机理的竞争反应路径,甚至略有优势。可以看出,B酸强度增加,分步机理和协同机理的反应活化能差异逐渐减少,说明B酸强度增强对分步机理更有利。B酸强度增强,甲醇脱水反应会更容易,生成乙烯的可能性更大,但是苯和乙烯的乙基化反应被抑制,乙苯的选择性不会因此提高7,8,14。实验中提高ZSM-5分子筛的硅铝比,苯的转化率和甲苯的选择性升高。
3.2 104T模型中苯和甲醇甲基化反应
3.2.1 分步机理
在分步机理中,甲醇脱水步骤的吸附物种和过渡态结构图如图5所示,主要的几何结构参数见表1,相对能量在反应能线图6中给出。在反应初期,在104T9和104T12模型活性位上,甲醇形成的吸附结构被标记为Ads1_Met和Ads2_Met,分别如图5(a)和图5(d)所示。在Ads1_Met和Ads2_Met结构中,甲醇的羟基氧(O3)原子和分子筛的B酸质子(H1)之间形成氢键,氢键的键长分别为0.143和 0.155 nm。可以看出,甲醇与104T9分子筛之间形成的氢键更稳定。甲醇的吸附能分别为-116.5和-109.9 kJ·mol-1,与Mazar等30用PBE+D方法计算得到的甲醇的吸附能相似(正弦孔道中:-118 kJ·mol-1;直孔道中:-132 kJ·mol-1),与实验结果(-115±5 kJ·mol-1)38相符合。比较甲醇的吸附能可知,甲醇在B酸酸性更强的104T9模型上吸附比在104T12模型上吸附更稳定。吸附后甲醇的C―O键分别伸长了0.003和0.002 nm,说明甲醇的C―O键强度被削弱,甲醇分子已经被活化,104T9模型中甲醇的C―O键被活化的程度更大。
分子筛B酸(H1)质子进攻甲醇的羟基氧(O3)原子导致C―O(C1―O3)键断裂,甲醇解离出甲基基团,释放一分子水。在过渡态结构(TS1_Met,图5 (b);TS2_Met,图5(e))中,甲基基团变为sp2杂化的甲基碳正离子结构,甲基碳正离子被分子筛骨架氧(O2)原子和O3原子共同稳定。在TS1_Met结构中,C1…O2和C1…O3的原子间距离分别为0.209和0.192 nm。在TS2_Met结构中,C1…O2和C1…O3的原子间距离分别为0.214和0.193 nm。与TS2_Met相比,TS1_Met中甲基碳正离子与O2和O3原子之间的距离更近,说明该结构中甲基碳正离子与氧原子之间的相互作用更强,TS1_Met比
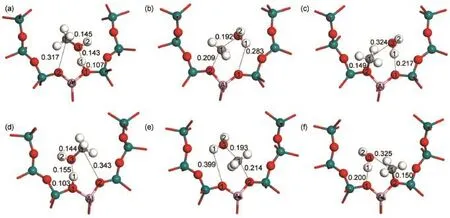
图5 104T模型中分步机理的甲醇脱水步骤的吸附物种和过渡态Fig.5 Adsorbed species and transition states in methanol dehydration step of the stepwise mechanism in the 104T models
表1 104T模型中苯和甲醇甲基化分步机理的主要物种的几何结构参数
Atom labels are given in Fig.5 and Fig.7.L is bond length andA is bond angle.
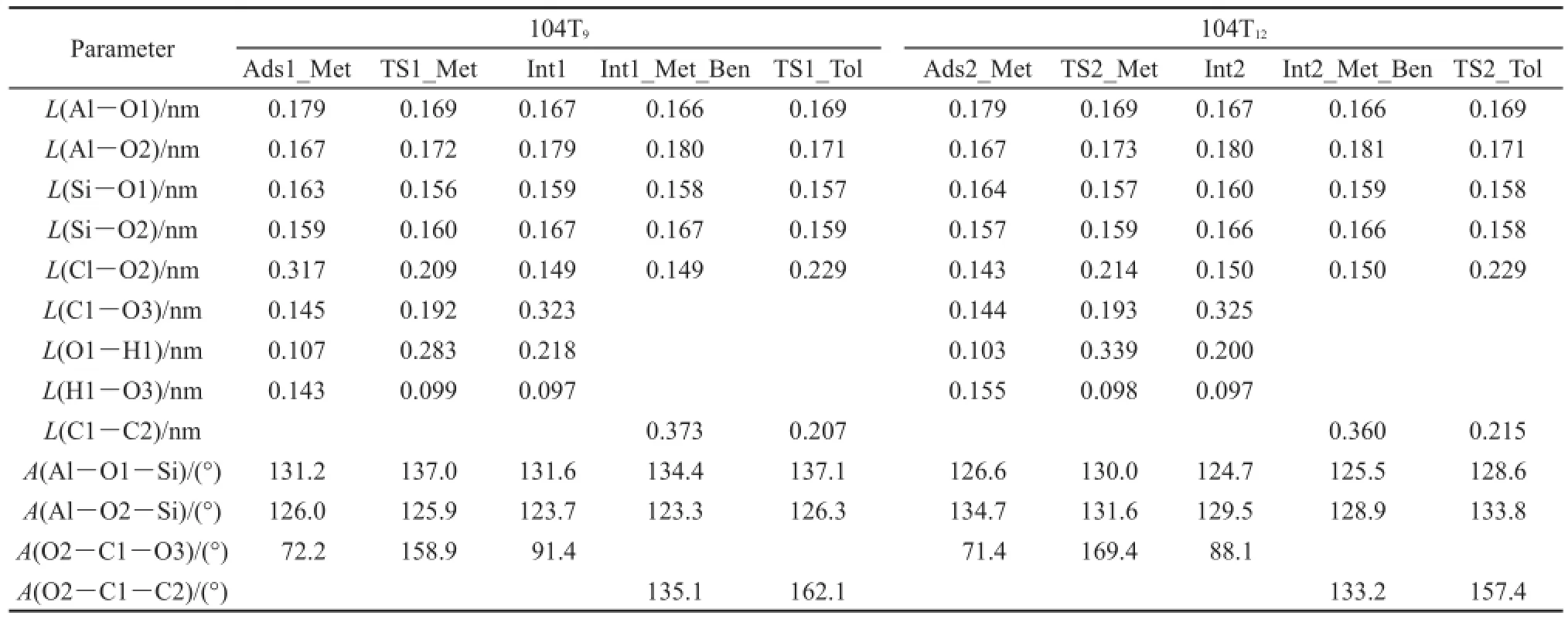
Table 1 Optimized geometric parameters of the key species involved in the stepwise mechanism of benzene methylation with methanol in the 104T models TS2_Met更稳定。在甲醇脱水产物结构(Int1,图5 (c);Int2,图5(f))中,甲基碳正离子和O2原子之间形成共价键相互作用,生成甲氧基中间体产物。产物水的H1原子和分子筛的初始碱性位(O1原子)之间形成氢键,水分子在甲氧基中间体附近吸附。最后,生成的水分子分别克服47.1和56.1 kJ·mol-1能垒,从活性位上脱附。甲氧基中间体分别命名为Int1_Met和Int2_Met。在104T9和104T12模型中,甲醇脱水步骤的反应活化能分别为143.6和161.9 kJ·mol-1(ΔEa=18.3 kJ·mol-1),反应分别吸热59.4和79.3 kJ·mol-1。比较甲醇脱水步骤的反应活化能可知,甲醇在104T9模型活性位上更容易发生脱水反应。
在甲基化步骤中,苯分子分别与Int1_Met和 Int2_Met以σ-π相互作用形成吸附反应物结构,Int1_Met_Ben和Int2_Met_Ben,分别如图7(a)和图7(c)所示。吸附能分别为-88.2和-91.9 kJ·mol-1。在甲基化反应的过渡态(TS1_Tol和TS2_Tol,图7 (b)和图7(d))结构中,甲氧基的甲基基团从分子筛骨架氧(O2)原子向苯环迁移,形成的甲基碳正离子被O2原子和苯环上的π电子共同稳定。TS1_Tol结构中C1…O2和C1…C2的距离依次为0.229和0.207 nm;TS2_Tol结构中C1…O2和C1…C2的距离依次为0.229和0.215 nm。经过甲基化过渡态,苯和甲氧基甲基化生成甲苯产物,质子回到分子筛最初的碱性位(O1原子)。苯和甲氧基甲基化生成甲苯步骤的活化能为86.6和89.7 kJ·mol-1。从104T9和104T12模型中苯和甲醇甲基化分步机理的反应能线图6可以看出,甲醇脱水为速率控制步骤,分步机理的总反应活化能分别为143.6和161.9 kJ·mol-1,104T9模型中苯分步甲基化反应活性更高。
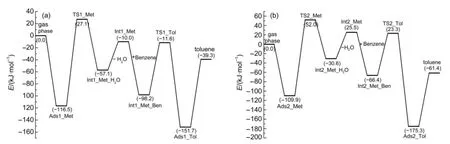
图6 (a)104T9模型和(b)104T12模型中苯和甲醇甲基化分步机理的反应能线图Fig.6 Reaction energy profile for the stepwise mechanism of benzene methylation with methanol to form toluene in (a)104T9model and(b)104T12model
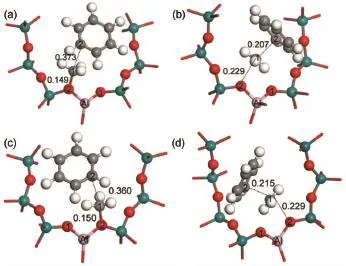
图7 104T模型中分步机理的甲基化步骤中吸附物种和过渡态Fig.7 Adsorbed species and transition states in the methylation step of the stepwise mechanism in the 104T models
3.2.2 协同机理
在协同机理中,在甲基化反应初期,一分子苯与甲醇在104T9和104T12分子筛活性位上形成共吸附反应物结构,分别被标记为Co_ads1和Co_ads2,如图8(a)和图8(c)所示。苯和甲醇共吸附的吸附能分别为-180.4和-173.3 kJ·mol-1。在共吸附反应物结构中,甲醇的羟基氧原子和B酸质子形成氢键(O3―H1),甲醇的羟基氢(H2)原子和苯环的π电子之间形成H-π相互作用。在Co_ads1结构中,分子筛桥羟基(H1―O1)键长为0.102 nm,H1…O3原子间距离为0.163 nm,C1…C2原子间距离为0.365 nm。在Co_ads2结构中,H1―O1键长为0.100 nm,H1…O3原子间距离为0.176 nm,C1…C2原子间距离为0.378 nm。比较几何结构参数可以看出,在104T9模型中,B酸质子与吸附物种之间以及甲醇与苯之间的相互作用更强。苯和甲醇沿着协同机理发生甲基化反应的能线图如图9所示,主要物种的结构参数在表2中给出。
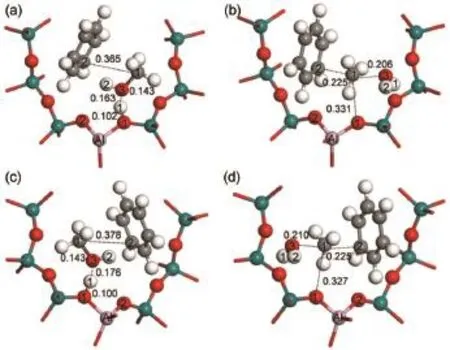
图8 104T模型中协同机理的甲基化步骤的吸附物种和过渡态Fig.8 Adsorbed species and transition states in the concerted mechanism in the 104T models
分子筛B酸质子从分子筛碱性氧原子向甲醇迁移的行为诱导苯和甲醇沿着协同机理的反应路径发生甲基化反应。在104T9和104T12模型中,协同机理的甲基化过渡态结构如图8(b)和图8(d)所示,结构分别被命名为TS1_Tol和TS2_Tol。在甲基化过渡态结构中,甲醇的碳-氧键已经断裂,解离的羟基和B酸质子生成一分子水,甲基基团变成甲基碳正离子被苯环的π电子和生成的水分子共同稳定。在TS1_Tol结构中,O3…C1和C1…C2原子间距离分别为0.206和0.225 nm。在TS2_Tol结构中,O3…C1和C1…C2原子间距离分别为0.210和0.225 nm。可以看出TS1_Tol和TS2_Tol的键长结构参数相近,前者甲基碳正离子与水分子之间的距离更近一些,结构更稳定。在苯和甲醇协同甲基化过程中,苯环的C2原子快速地将氢相连的质子,氢质子回到分子筛骨架,完成了甲基化反应,最终生成甲苯产物和一分子水。在104T9和104T12模型中,苯和甲醇沿着协同机理进行甲基化反应的总反应活化能分别为137.5和148.0 kJ· mol-1,104T9模型中苯协同甲基化反应活性更高。
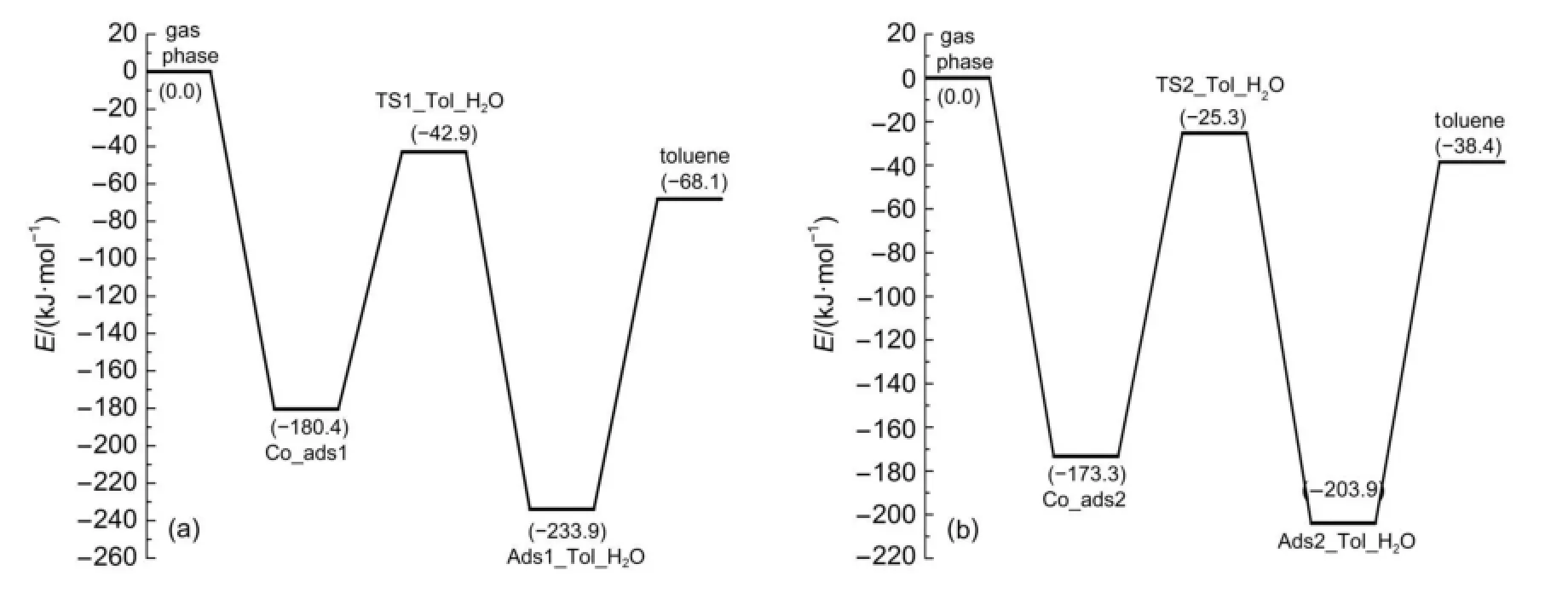
图9 (a)104T9模型和(b)104T12模型中苯和甲醇甲基化协同机理的反应能线图Fig.9 Reaction energy profile for the concerted mechanism of benzene methylation with methanol to form toluene in (a)the 104T9model and(b)the 104T12model
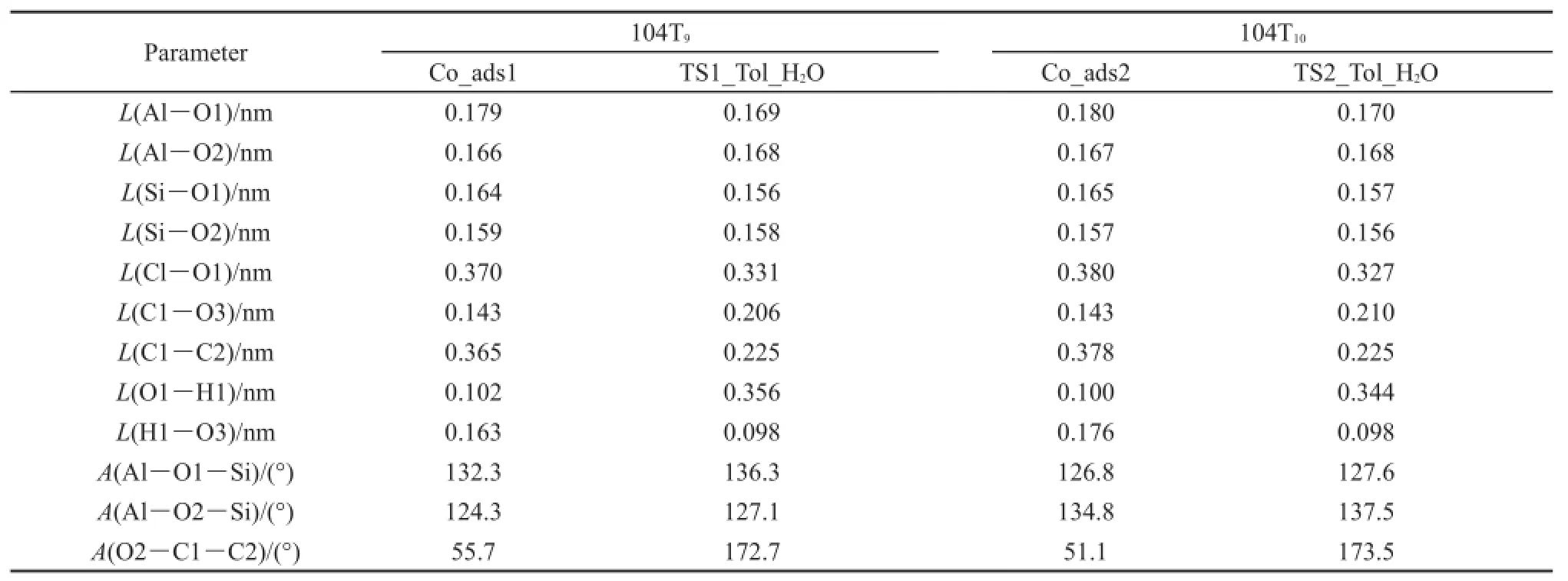
表2 104T模型中苯和甲醇甲基化协同机理的主要物种的几何结构参数Table 2 Optimized geometric parameters of the key species involved in the concerted mechanism of benzene methylation with methanol in the 104T models
进一步计算了104T9和104T12模型的PA和NH3吸附能,104T9模型的PA比104T12模型的PA高29.2 kJ·mol-1。氨气在104T9模型活性位上的吸附能比在104T12模型活性位上的吸附能低55.3 kJ· mol-1。104T9H-ZSM-5分子筛活性位上B酸酸性更强。在104T12分子筛模型中,苯和甲醇甲基化的分步机理和协同机理的反应活化能分别为161.9和148.0 kJ·mol-1。分步机理的反应活化能比协同机理高13.9 kJ·mol-1,说明协同机理为主导反应路径。在104T9分子筛模型中,苯和甲醇甲基化的分步机理和协同机理的反应活化能分别为143.6和137.5 kJ·mol-1。分步机理和协同机理的反应活化能相差6.1 kJ·mol-1,两种反应机理的反应活化能差异不大,为竞争反应路径。可以得出结论,HZSM-5分子筛B酸酸性越强,对苯和甲醇甲基化反应的催化活性越好。随着B酸酸性增强,由于分步机理的反应活化能降低的幅度比协同机理的反应活化能降低的幅度更大。在分步机理中,甲醇脱水生成的甲氧基中间体的氢原子与分子筛活性位的碱性氧原子之间存在相互作用。实验研究表明当反应温度超过493 K时,甲氧基中间体可以脱质子生成内鎓盐或卡宾物种,进而聚合成大体积的稠环芳烃物种导致积炭4,39。结合理论计算结果可知,当分步机理成为苯甲基化反应的主导反应路径时,甲氧基在正弦形孔道和直孔道的交叉处生成,大体积稠环芳烃会在交叉处堵塞分子筛孔道,进而阻碍甲苯产物从孔内向孔外的扩散过程,甲苯选择性也会因此受到影响。除此之外,大体积稠环芳烃一旦生成很难从活性位上脱附,因此覆盖了活性位,导致催化剂稳定性和活性都变差。因此,调变ZSM-5催化剂B酸强度,促使苯和甲醇沿着协同机理发生甲基化反应,可以提高催化剂的稳定性和催化性能。
4 结论
采用ωB97X-D/6-31G(d,p)//ONIOM2(B3LYP/6-31G(d,p):UFF)方法,计算了5T、12T和104T HZSM-5催化剂模型中苯和甲醇甲基化分步反应机理和协同反应机理。考察了催化剂B酸强度对苯和甲醇甲基化反应机理的影响。计算结果表明,HZSM-5分子筛B酸酸性越强,苯和甲醇甲基化反应活化能越低,分子筛对苯和甲醇甲基化反应的催化活性越好。催化剂的B酸强度对苯甲基化主导反应路径有重要的影响,B酸强度不同,苯甲基化反应的主导反应路径可能不同。随着B酸酸性增强,与协同机理相比,分步机理的反应活化能降低的幅度更大,证明了B酸酸性增强对分步机理更有利。当H-ZSM-5分子筛催化苯和甲醇甲基化的分步机理成为主导反应路径时,分步机理中甲醇经过脱水步骤生成甲氧基中间体,其解离并进一步聚合成大体积积炭物种的副反应会导致分子筛的稳定性下降。理论计算结果表明,在一定范围内调试H-ZSM-5分子筛催化剂B酸强度,促使苯和甲醇沿着协同机理的反应路径进行甲基化反应,同时增加酸强度,可以提高催化剂的稳定性和催化活性。
(1) Hu,H.M.Methylation of Benzene with Methanol over a Series of ZSM-5 Catalysts.M.S.Dissertation.Human Normal University,Changsha,2007.[胡慧敏.ZSM-5系列分子筛催化剂上苯与甲醇的烷基化反应研究[D].长沙:湖南师范大学, 2007.]
(2) Liu,J.Chem.Prod.Tech.2011,18,19.[刘 键.化工生产与技术,2011,18,19.]
(3) Li,Y.Y.Catalysts Research forAlkylation of Benzene with Methanol.M.E.Dissertation.East China University of Science and Technology,Shanghai,2011.[李燕燕.苯、甲醇烷基化催化剂的研究[D].上海:华东理工大学,2011.]
(4)Yuan,P.;Wang,H.;Xue,Y.F.;Li,Y.C.;Wang,K.;Dong,M.; Fan,W.B.;Qin,Z.F.;Wang,J.G.Acta Phys.-Chim.Sin. 2016,32,1775.[袁 苹,王 浩,薛彦峰,李艳春,王 凯,董 梅,樊卫斌,秦张峰,王建国.物理化学学报,2016,32, 1775.]doi:10.3866/PKU.WHXB201604141
(5)Zhao,B.;Liu,M.;Tan,W.;Wu,H.Y.;Guo,X.W.Acta Petro. Sin.2013,29,605.[赵 博,刘 民,谭 伟,吴宏宇,郭新闻.石油学报,2013,29,605.]
(6)Liu,Y.;Li,D.F.;Wang,T.Y.;Liu,Y.;Xu,T.;Zhang,Y.ACS Catal.2016,6,5366.doi:10.1021/acscatal.6b01362
(7)Xu,Y.R.;Xu,X.L.;Zhu,X.D.Chem.React.Eng.Technol. 2015,31,475.[徐亚荣,徐新良,朱学栋.化学反应工程与工艺,2015,31,475.]
(8) Hu,H.L.;Lyu,J.H.;Rui,J.Y.Catal.Sci.Technol.2016,6, 2647.doi:10.1039/c5cy01976a
(9) Svelle,S.;Bjørgen,M.J.Phys.Chem.A 2010,114,12548.doi: 10.1021/jp108892e
(10) Rakoczy,J.;Romotowski,T.Zeolites 1993,13,256. doi:10.1016/0144-2449(93)90003-L
(11) Svelle,S.;Visur,M.;Olsbye,U.;Saepurahman;Bjørgen,M. Top Catal.2011,54,897.doi:10.1007/s11244-011-9697-7
(12) Vos,A.M.;Nulens,H.L.;De Proft,F.;Schoonheydt,R.A.; Geerlings,P.J.Phys.Chem.B 2002,106,2026.doi:10.1021/ jp014015a
(13) Van der Mynsbrugge,J.;Visur,M.;Olsbye,U.;Beato,P.; Bjørgen,M.;Van Speybroeck,V.;Svelle,S.J.Catal.2012,292, 201.doi:10.1016/j.jcat.2012.05.015
(14) Hansen,N.;Brüggemann,T.;Bell,T.A.;Keil,J.F.J.Phys. Chem.C 2008,112,15402.doi:10.1021/jp8036022
(15) Olson,D.H.;Kokotailo,G.T.;Lawton,S.L.;Meier,W.M. J.Phys.Chem.1981,85,2238.doi:10.1021/j150615a020
(16)Kokotailo,G.T.;Lawton,S.L.;Olson,D.H.;Meier,W.M. Nature 1978,272,437.doi:10.1038/272437a0
(17) Vankoningsveld,H.;Vanbekkum,H.;Jansen,J.C.Acta Crystallogr.Sect.B,Struct.Sci.1987,43,127.doi:10.1107/ S0108768187098173
(18) Chatterjee,A.;Vetrivel,R.Microporous Mater.1994,3,211. doi:10.1016/0927-6513(94)00032-8
(19)Nie,X.W.;Janik,M.J.;Guo,X.W.;Song,C.S.J.Phys.Chem. C 2012,116,4071.doi:10.1021/jp209337m
(20)Li,L.L.;Nie,X.W.;Song,C.S.;Guo,X.W.Acta Phys.-Chim. Sin.2013,29,754.[李玲玲,聂小娃,宋春山,郭新闻.物理化学学报,2013,29,754.]doi:10.3866/PKU.WHXB201302063
(21)Li,L.L.;Janik,J.M.;Nie,X.W.;Song,C.S.;Guo,X.W.Acta Phys.-Chim.Sin.2015,31,56.[李玲玲,Janik,J.Michael,聂小娃,宋春山,郭新闻.物理化学学报,2015,31,56.] doi:10.3866/PKU.WHXB201411052
(22) Fripiat,J.G.;Berger-André,F.;André,J.M.;Derouane,E.G. Zeolites 1983,3,306.doi:10.1016/0144-2449(83)90174-4
(23) Derouane,E.G.;Fripiat,J.G.Zeolites 1985,5,165. doi:10.1016/0144-2449(85)90025-9
(24) Jungsuttiwong,S.;Lomratsiri,J.;Limtrakul,J.Int.J.Quant. Chem.2011,111,2275.doi:10.1002/qua.v111.10
(25)Maseras,F.;Morokuma,K.J.Comput.Chem.1995,16,1170. doi:10.1002/jcc.540160911
(26)Dapprich,S.;Komáromi,I.;Byun,K.S.;Morokuma,K.; Frisch,M.J.J.Mol.Struct.-Theochem 1999,462,1.doi:10.1016/S0166-1280(98)00475-8
(27) Matsubra,T.;Sieber,S.;Morokuma,K.Int.J.Quant.Chem. 1996,60,1101.doi:10.1002/(SICI)1097-461X(1996)60:6〈1101::AID-QUA1>3.0.CO;2-3
(28)Rappe,A.K.;Upton,T.H.J.Am.Chem.Soc.1992,114,7507. doi:10.1021/ja00045a026
(29)Vreven,T.;Morokuma,K.J.Comput.Chem.2000,21,1419. doi:10.1002/1096-987X(200012)21:16〈1419::AID-JCC1>3.0. CO;2-C
(30)Mazar,M.N.;Al-Hashimi,S.;Bhan,A.;Cococcioni,M. J.Phys.Chem.C 2012,116,19385.doi:10.1021/jp306003e
(31) Van der Mynsbrugge,J.;Hemelsoet,K.;Vandichel,M.; Waroquier,M.;Van Speybroeck,V.J.Phys.Chem.C 2012,116, 5499.doi:10.1021/jp2123828
(32)Chai,J.D.;Head-Gordon,M.Phys.Chem.Chem.Phys.2008, 10,6615.doi:10.1039/b810189b
(33)Goerigk,L.;Grimme,S.Phys.Chem.Chem.Phys.2011,13, 6670.doi:10.1039/c0cp02984j
(34) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, RevisionA.02;Gaussian,Inc.:Pittsburgh,PA,2009.
(35) Nie,X.W.;Janik,J.M.;Guo,X.W.;Liu,X.;Song,C.S.Catal. Today 2011,165,120.doi:10.1016/j.cattod.2010.11.070
(36) Corma,A.;Llopis,F.;Viruela,P.;Zicovichwilson,C.J.Am. Chem.Soc.1994,116,134.doi:10.1021/ja00080a016
(37) Kondo,J.N.;Yamazaki,H.;Yokoi,T.;Tatsumi,T.Catal.Sci. Technol.2015,5,3598.doi:10.1039/c5cy00600g
(38) Lee,C.C.;Gorte,R.J.;Farneth,W.E.J.Phys.Chem.B 1997, 101,3811.doi:10.1021/jp970711s
(39)Wang,W.;Buchholz,A.;Seiler,M.;Hunger,M.J.Am.Chem. Soc.2003,125,15260.doi:10.1021/ja0304244
Reaction Mechanism of Benzene Methylation with Methanol over H-ZSM-5 Catalyst
LI Ling-Ling1CHEN Ren1DAI Jian1SUN Ye1ZHANG Zuo-Liang1LI Xiao-Liang1NIE Xiao-Wa2,*SONG Chun-Shan2,3GUO Xin-Wen2,*
(1Department of Metallurgical Engineering,Liaoning Institute of Science and Technology,Benxi 117004,Liaoning Province, P.R.China; 2State Key Laboratory of Fine Chemicals,PSU-DUT Joint Center for Energy Research,School of Chemical Engineering,Dalian University of Technology,Dalian 116024,Liaoning Province,P.R.China; 3EMS Energy Institute, PSU-DUT Joint Center for Energy Research and Department of Energy&Mineral Engineering, Pennsylvania State University,University Park,PA 16802,USA)
The stepwise and concerted mechanisms of benzene methylation with methanol were studied with the 5T,12T,104T9,and 104T12H-ZSM-5 models using the“our own-N-layered integrated molecular orbital+ molecular mechanics”(ONIOM)in combination with density functional theory(DFT)methods.The structures of intermediate species and transition states were described.The effect of the Brønsted(B)acid strength of HZSM-5 catalyst on the reaction mechanism of benzene methylation with methanol was considered.The reactionactivation energy results indicate that benzene methylation with methanol preferentially occurs over H-ZSM-5 catalyst with greater B acid strength,and a lowering of the activation barrier was observed.With increasing B acid strength,the reaction activation energy of the stepwise mechanism decreases more than that of the concerted mechanism.Increasing the B acidic strength is more beneficial to the stepwise mechanism.When the stepwise mechanism becomes the dominant reaction path,the secondary reaction arising from further formation of bulky hydrocarbons through the methoxide intermediate produced in the methanol dehydration step of the stepwise mechanism might lead to the inactivation of the H-ZSM-5 catalyst owing to coke formation. Reasonable modulation the acid strength of the H-ZSM-5 catalyst is important in improving its catalytic activity and stability of the catalyst.
Density functional theory;ONIOM;Benzene methylation;Methanol;H-ZSM-5
O641
10.3866/PKU.WHXB201612162
Received:October 31,2016;Revised:December 16,2016;Published online:December 16,2016.
*Corresponding authors.GUO Xin-Wen,Email:guoxw@dlut.edu.cn;Tel:+86-411-84986133.NIE Xiao-Wa,Email:niexiaowa@dlut.edu.cn;
Tel:+86-411-84986486.
The project was supported by the Scientific Research Foundation for the general Program of Department of Education of Liaoning Province of China (L2014503),Natural Science Foundation of Liaoning Province,China(201602403),Research Fund for the Doctoral Program of Liaoning Institute of Science and Technology,China(1406B08)and National Natural Science Foundation of China(21503027).辽宁省教育厅科
学研究一般项目基金(L2014503),辽宁省自然科学基金项目(201602403),辽宁科技学院博士科研启动基金项目(1406B08)及国家自然科学基金项目(21503027)资助
