固相萃取-超高效液相色谱-串联质谱法测定乳粉和果冻中的氯化胆碱
2017-05-11李晰晖黄小贝李永强
汪 辉, 刘 江, 李晰晖, 曹 阳, 黄小贝, 李永强
(长沙市食品药品检验所, 湖南省食品安全生产工程技术研究中心, 湖南 长沙 410013)
技术与应用
固相萃取-超高效液相色谱-串联质谱法测定乳粉和果冻中的氯化胆碱
汪 辉*, 刘 江, 李晰晖, 曹 阳, 黄小贝, 李永强
(长沙市食品药品检验所, 湖南省食品安全生产工程技术研究中心, 湖南 长沙 410013)
建立了固相萃取-超高效液相色谱-串联质谱测定乳粉和果冻中氯化胆碱的分析方法。样品在10 mL 0.02 mol/L乙酸铵溶液(冰乙酸调节pH至3.0)中水解3 h后离心,上清液经DIKMA ProElut PLS固相萃取柱(60 mg/3 mL)净化后,用Agilent ZORBAX 300 SCX色谱柱(150 mm×2.1 mm, 5 μm)进行分离,通过电喷雾正离子(ESI+)模式电离,多反应监测(MRM)模式进行定性和定量分析。方法的检出限(LOD,S/N=3)和定量限(LOQ,S/N=10)分别为0.15 mg/kg和0.50 mg/kg,加标回收率为70.8%~100.2%,相对标准偏差(RSD)均不大于6.83%(n=6)。目标化合物在0.05~8.0 mg/L范围内线性关系良好,线性方程为Y=2.05×105X+3.24×104,相关系数(r)为0.996。对市售乳粉和果冻中的氯化胆碱进行检测,结果表明,乳粉和果冻中氯化胆碱的含量分别为251.0~2 448 mg/kg和0.261~0.314 mg/kg。该法准确可靠、灵敏度高,适用于乳粉和果冻中氯化胆碱的测定。
超高效液相色谱-串联质谱;固相萃取;氯化胆碱;乳粉;果冻
氯化胆碱(choline chloride)是胆碱的衍生物,具有胆碱的生物活性[1],是动物生长过程中不可缺少的一种水溶性维生素,作为一种饲料添加剂,已广泛应用于动物饲料工业中[2-4]。生物体内胆碱的缺乏可能会引起发育受阻、生长缓慢、繁殖能力差等不良生理现象[5,6]。GB 14880-2012《食品营养强化剂使用标准》中规定氯化胆碱可代替胆碱,作为营养强化剂添加至调制乳粉和果冻中。但食品用氯化胆碱目前无相关的国家标准对其含量、水分、灼烧残渣等理化指标做出限定,不同厂家的产品中氯化胆碱的含量高低不一,导致一些企业生产的食品中氯化胆碱的含量达不到国家标准[3,4]。2016年7月,我国发布了《食品营养强化剂氯化胆碱》的征求意见稿,意味着食品级氯化胆碱即将规范化,并在食品工业中广泛应用。因此,建立食品中氯化胆碱的分析方法,有利于食品生产企业的质量控制,也为监管部门提供必要的技术支撑。
氯化胆碱沸点高、热稳定性好,其化学结构既无紫外吸收基团,又无荧光特性,无法利用气相色谱法、高效液相色谱-紫外/荧光法进行检测[6]。目前,氯化胆碱的分析方法主要有离子色谱法[1,7-10]、分光光度法[4,11]、雷氏重量法[12,13]、定氮法[14]、高效液相色谱-蒸发光散射法[15]和液相色谱-串联质谱法[5,16]。离子色谱法采用离子交换柱分离,有一定的选择性,但电导检测器是通用型检测器,灵敏度较低;分光光度法和常规分析法的实验操作繁琐、干扰大,仅适用于常量分析。液相色谱-串联质谱法既能对目标化合物进行有效分离,又能弥补其他检测器的缺陷,专一性强,灵敏度高。固相萃取技术能够对样品进行净化,去除部分干扰物,有效地降低基质效应。本文建立了固相萃取-超高效液相色谱-串联质谱(UPLC-MS/MS)测定乳粉和果冻中氯化胆碱的分析方法。采用亲水亲脂固相萃取柱对样品进行净化,并采用强阳离子交换(SCX)色谱柱进行分离,通过电喷雾正离子模式(ESI+)电离,多反应监测(MRM)模式扫描,实现了乳粉和果冻中氯化胆碱的定性和定量分析,结果令人满意。
1 实验部分
1.1 仪器与试剂
Agilent 1290超高效液相色谱仪、Agilent 6460三重四极杆质谱仪(美国Agilent公司); DIKMA ProElut PLS固相萃取柱(60 mg/3 mL); 24位固相萃取装置(美国Supelco公司); DK-98-Ⅱ电热恒温水浴锅(天津市泰斯特仪器有限公司); CT14D型台式高速离心机(上海天美生化仪器设备工程有限公司); Milli-Q超纯水器(美国Millipore公司)。
婴儿配方乳粉质控样品(批号QC-IP-019,胆碱指定值165.8 mg/100 g,满意值范围为149.0~182.6 mg/100 g,中国检验检疫科学研究院测试评价中心);甲醇、乙腈为色谱纯(北京DIKMA公司);冰乙酸为色谱纯(天津化学试剂研究所);乙酸铵、氯化胆碱为分析纯(国药集团化学试剂有限公司)。
准确称取适量的氯化胆碱,用甲醇溶解并定容,配制质量浓度为1.0 g/L的标准储备液,于-18 ℃保存;根据实验要求逐级稀释标准储备液至适当浓度,配制标准工作液,现用现配。
1.2 UPLC-MS/MS条件
色谱柱:Agilent ZORBAX 300 SCX色谱柱(150 mm×2.1 mm, 5 μm)(美国Agilent公司);柱温:30 ℃;流动相:0.02 mol/L乙酸铵(冰乙酸调节pH至3.0)-乙腈(60∶40, v/v);等度洗脱;流速:0.4 mL/min;进样量:1 μL。
离子源:ESI源;正离子模式;毛细管电压:4 000 V;雾化器(N2)压力:3.4×105Pa;干燥气(N2)温度:350 ℃;干燥气(N2)流速:10 L/min;裂解电压:105 V;扫描模式:多反应监测(MRM);母离子:m/z104;定量子离子:m/z60,碰撞能量:15 eV;定性子离子:m/z45,碰撞能量:20 eV。
1.3 样品前处理
准确称量1.0 g乳粉或果冻于25 mL离心管中,加入10 mL 0.02 mol/L乙酸铵溶液(冰乙酸调节pH至3.0),混匀,置于70 ℃水浴中水解3 h,冷却,摇匀,以10 000 r/min离心5 min,取上清液待用。分别用3 mL甲醇、3 mL超纯水活化DIKMA ProElut PLS固相萃取柱(60 mg/3mL),移取3 mL上清液上样,收集滤液,供LC-MS/MS分析。
2 结果与讨论
2.1 色谱条件的优化
2.1.1 色谱柱的选择
本实验考察了Agilent SB-C18色谱柱(150 mm×2.1 mm, 5 μm)和Agilent SCX色谱柱(150 mm×2.1 mm, 5 μm)对目标化合物分离效果的影响。结果表明,采用Agilent SB-C18色谱柱时,以0.02 mol/L乙酸铵(冰乙酸调节pH至3.0)-乙腈为流动相,氯化胆碱在水相比例较高时,几乎无保留,而随着水相比例降低,保留时间增大,但色谱峰拖尾严重,采用梯度洗脱,效果仍不令人满意;采用Agilent SCX色谱柱时,因氯化胆碱主要以离子形式存在于水溶液中,其分离效果良好,色谱峰峰形尖锐,分离度高。因此选择Agilent SCX色谱柱(150 mm×2.1 mm, 5 μm)分离氯化胆碱。
2.1.2 流动相的选择
考察了流动相中不同浓度(0.01、0.015、0.02、0.025 mol/L)的乙酸铵对目标化合物分离效果的影响(见图1)。结果发现,目标化合物的保留时间随乙酸铵浓度的增大而减小,响应强度随乙酸铵浓度的增大而增大。但是过高浓度的乙酸铵与有机相混合时可能会析出,从而堵塞色谱柱,因此乙酸铵的浓度不宜过高。结合目标化合物的响应强度和保留时间,最终选择浓度为0.02 mol/L的乙酸铵(见图1b)作为流动相中的水相。
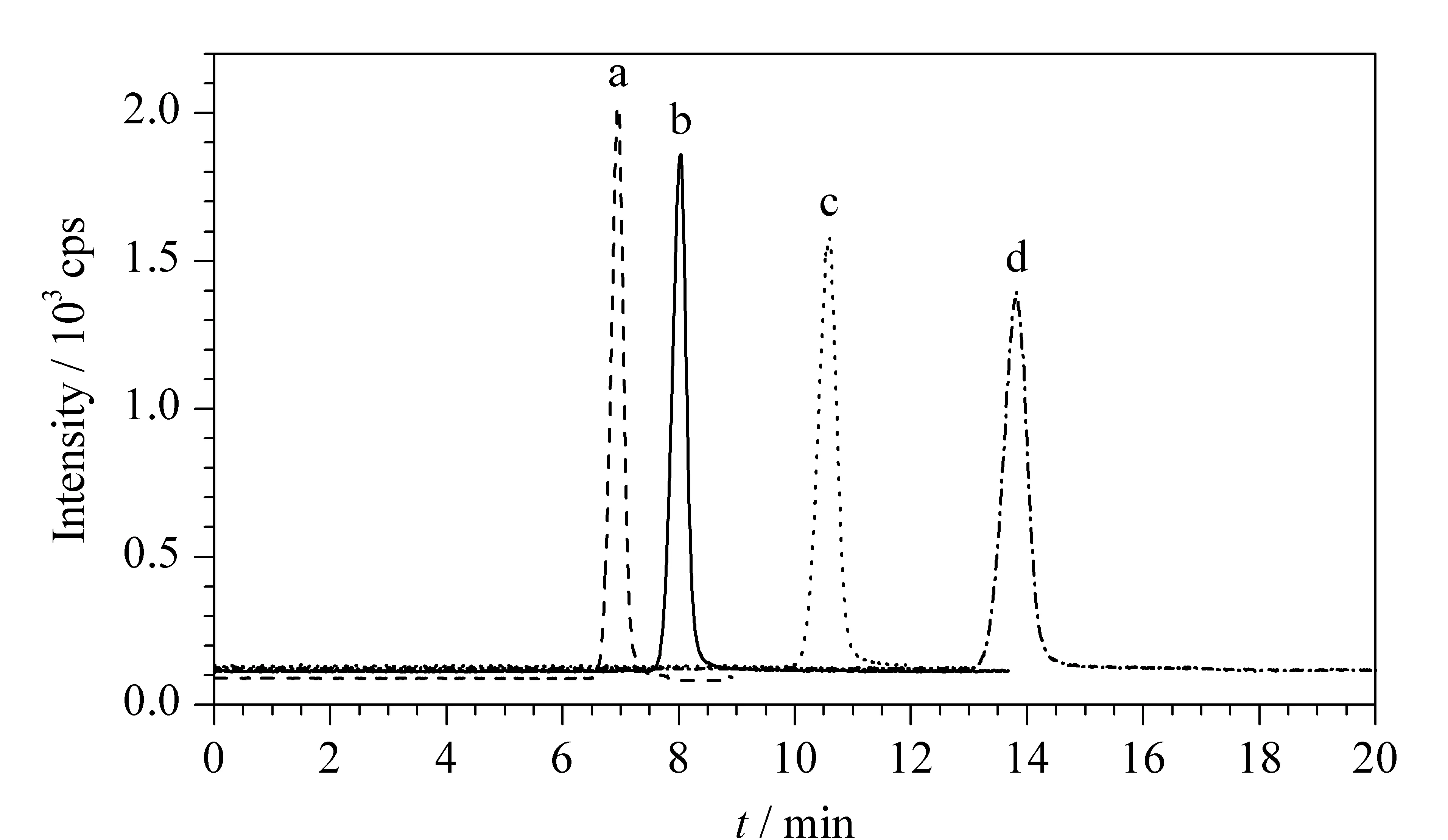
图 1 采用不同浓度的乙酸铵时氯化胆碱的色谱图Fig. 1 Chromatograms of choline chloride by different concentrations of ammonium acetatea. 0.025 mol/L; b. 0.02 mol/L; c. 0.015 mol/L; d. 0.01 mol/L.
考察了不同pH值(6.8、4.5和3.0,用冰乙酸调节)的乙酸铵对目标化合物分离效果的影响(见图2)。结果发现,当乙酸铵的pH值为3.0时,氯化胆碱的响应强度最高(见图2a)。因此最终选择将乙酸铵的pH调节至3.0。

图 2 采用不同pH值的乙酸铵时氯化胆碱的色谱图Fig. 2 Chromatograms of choline chloride under different pH values of ammonium acetatepH values of ammonium acetate: a. 3.0; b. 4.5; c. 6.8.
考察了当0.02 mol/L乙酸铵(冰乙酸调节pH至3.0)-乙腈为流动相时,二者不同的体积比(70∶30、60∶40、50∶50、40∶60)对目标化合物分离效果的影响(见图3)。结果发现,目标化合物的保留时间随乙酸铵体积分数的增加而减小,而其响应强度先增大后减小。当流动相为0.02 mol/L乙酸铵(pH 3.0)-乙腈(60∶40, v/v)时,氯化胆碱的响应强度最大,色谱峰峰形对称(见图3b),因此选为实验所用。

图 3 采用不同比例的流动相时氯化胆碱的色谱图Fig. 3 Chromatograms of choline chloride by different ratios of mobile phaseVolume ratios of ammonium acetate (0.02 mol/L, pH 3.0) to acetonitrile: a. 70∶30; b. 60∶40; c. 50∶50; d. 40∶60.
2.2 质谱条件的优化
在不接色谱柱的情况下,采用直接进样的方式,将1.0 mg/L氯化胆碱标准溶液注入质谱离子源。根据目标化合物的结构和相对分子质量,在正离子模式下SIM扫描,得到目标化合物的分子离子峰,并通过连续测试得到优化的裂解电压。设定优化的裂解电压,以分子离子为母离子进行子离子扫描,得到碎片离子信息和优化的碰撞能量。按照欧盟2002/657/EC指令,对化合物进行定量确证必须达到4个确证点,即必须选择2个以上特征子离子。因此选择相对丰度较高的碎片离子作为定量特征离子,干扰较少的碎片离子作为定性特征离子(见图4),结合优化后的参数,采用MRM模式扫描,最终得到稳定性好、响应强度较高的色谱图。

图 4 氯化胆碱可能的裂解途径Fig. 4 Possible fragmentation pathways of the choline chloridem/z 60: quantitative ion; m/z 45: qualitative ion.
2.3 前处理条件的优化
分别考察了ProElut PWC(60 mg/3 mL)、ProElut PLS(60 mg/3 mL)、ProElut C18(60 mg/3 mL)、ProElut PLS(200 mg/6 mL)和ProElut C18(200 mg/6 mL)5种固相萃取柱对目标化合物净化效果的影响。ProElut PWC兼具弱阳离子交换保留和反相保留机理,专用于季铵盐类物质的保留;ProElut C18常被用来保留非极性和弱极性化合物,由于进行了封端处理,所以吸附剂的离子次级相互作用几乎不发挥作用;ProElut PLS对极性化合物和非极性化合物均有较好的保留。根据各固相萃取柱的特性,试验过程分别测试了每种固相萃取柱在上样、淋洗和洗脱3个步骤中目标化合物的回收率(见表1)。其中ProElut PWC固相萃取柱依次采用3 mL 5%(v/v)氨水、甲醇淋洗,3 mL含2%(v/v)甲酸的甲醇溶液洗脱;ProElut C18和PLS固相萃取柱采用3 mL 10%(v/v)甲醇水溶液淋洗,3 mL甲醇洗脱。结果表明,采用ProElut PWC(60 mg/3 mL)和ProElut PLS(200 mg/6 mL)固相萃取柱时,每个步骤回收率均不理想,其他3种固相萃取柱在上样过程的回收率均大于90%,且采用ProElut PLS(60 mg/3 mL)和ProElut C18(60 mg/3 mL)固相萃取柱时,回收率均在99%左右。考虑到ProElut PLS固相萃取柱兼具亲水基团和疏水基团,对极性化合物和非极性化合物均有较好保留,因此最终选用ProElut PLS(60 mg/3 mL)固相萃取柱对样品进行净化。
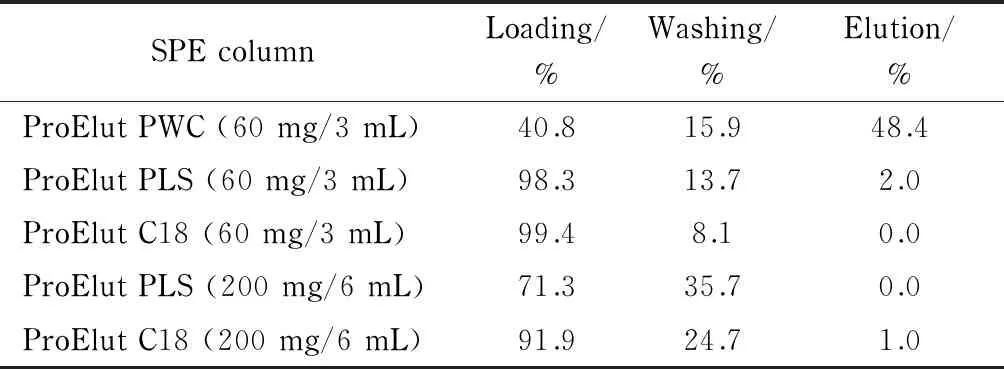
表 1 5种固相萃取柱在上样、淋洗和洗脱过程中
ProElut PWC column: washed by 3 mL 5%(v/v) ammonia water and 3 mL methanol, eluted by 3 mL methanol containing 2%(v/v) formic acid; ProElut C18 and PLS column: washed by 3 mL 10%(v/v) methanol aqueous solution, eluted by 3 mL methanol.
2.4 基质效应
采用LC-MS/MS分析时,由于样品溶液中的干扰物影响目标化合物的电离,导致其响应强度增强或减弱的现象称为基质效应(ME)[17,18]。为尽量降低基质效应,本实验采用缓冲溶液(乙酸铵-冰乙酸)提取样品,以减小溶液pH的变化;采用固相萃取净化,以减小过多干扰物的影响;采用较低的进样量,以减小高含量共流出组分的影响;采用阳离子交换柱分离,以减小相似极性化合物的影响。分别配制高、低两个水平的标准溶液和样品基质溶液,根据同一水平下样品基质溶液和标准溶液中氯化胆碱响应强度的比值,考察方法的基质效应[19],每个水平重复测定6次。结果表明,乳粉和果冻样品的基质效应分别为93.7%~98.2%和96.3%~103.8%(见表2)。因此本实验可忽略基质效应对测定结果的影响。

表 2 乳粉和果冻样品溶液的基质效应(n=6)
2.5 方法学考察
2.5.1 检出限、定量限和线性范围
在优化的色谱和质谱条件下,以3倍和10倍信噪比计算方法的检出限和定量限,分别为0.15 mg/kg和0.50 mg/kg。将氯化胆碱标准溶液用0.02 mol/L乙酸铵(冰乙酸调节pH至3.0)溶液逐级稀释,以氯化胆碱的峰面积为纵坐标(Y)、对应的质量浓度为横坐标(X, mg/L)绘制标准曲线。目标化合物在0.05~8.0 mg/L范围内线性关系良好,线性方程为Y=2.05×105X+3.24×104,相关系数(r)为0.996。
2.5.2 回收率和精密度
为验证方法的实用性,参考一般乳粉和果冻中目标化合物的含量,进行了加标回收率试验。分别在乳粉样品中添加100、200、2 000 mg/kg的氯化胆碱,在果冻样品中添加0.5、1.0、5.0 mg/kg的氯化胆碱,同一添加水平分别制备6份样品,供LC-MS/MS测试。结果表明,乳粉中氯化胆碱的加标回收率为80.8%~91.2%, RSD为2.04%~2.80%;果冻中氯化胆碱的加标回收率为70.8%~100.2%, RSD为2.39%~6.83%(见表3),可以满足日常检测需求。
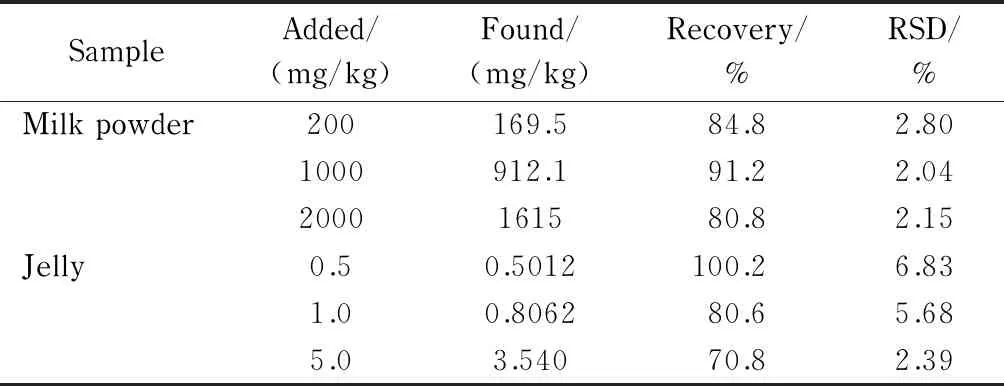
表 3 乳粉和果冻中氯化胆碱的加标回收率和相对标准偏差(n=6)
2.6 稳定性
随机抽取一份乳粉样品和果冻样品,对其进行前处理,随后分别在0、2、4、8、12 h测定氯化胆碱的含量,考察方法的稳定性。乳粉样品和果冻样品中氯化胆碱峰面积的RSD分别为4.81%和4.51%(n=5)。说明样品提取溶液在12 h内稳定性良好。

图 5 乳粉质控样品、实际乳粉和果冻样品中氯化胆碱的MRM色谱图Fig. 5 MRM chromatograms of choline chloride in a milk powder quality-control sample, a real milk powder and jelly sample
2.7 实际样品测定
采用本方法对乳粉质控样品(批号QC-IP-019)和市售的5个乳粉样品、5个果冻样品中氯化胆碱的含量进行测定,乳粉质控样品、实际乳粉和果冻样品的MRM色谱图见图5。乳粉质控样品中氯化胆碱(以胆碱计)的含量为156.7 mg/100 g,在满意值范围内,说明方法准确、可靠。10个市售样品均检出目标化合物,乳粉和果冻样品中氯化胆碱的含量分别为251.0~2 448 mg/kg和0.261~0.314 mg/kg。
3 结论
本文建立了固相萃取-超高效液相色谱-串联质谱测定乳粉和果冻中氯化胆碱含量的分析方法。该方法稳定性好、灵敏度高,可满足乳粉和果冻中氯化胆碱含量的测定,为今后企业对原料把关和食品监督部门的日常监管提供技术支撑。
[1] Cao W J, Cui H, Shen B Z, et al. Modern Scientific Instruments, 2012(6): 162
曹文军, 崔晗, 沈葆真, 等. 现代科学仪器, 2012(6): 162
[2] HG/T 2941-2004
[3] Zhong H J, Wei H. Feed Review, 2003(11): 20
钟红舰, 魏红. 饲料博览, 2003(11): 20[4] Chang B Y, Song R, Ding Y S, et al. China Feed, 2002(9): 23
常碧影, 宋荣, 丁永胜, 等. 中国饲料, 2002(9): 23
[5] Huang T, Tao B H, Chen Q. et al. Journal of Food Safety and Quality, 2014, 5(7): 2059
黄焘, 陶保华, 陈启, 等. 食品安全质量检测学报, 2014, 5(7): 2059
[6] Ding Y S, Mou S F. Chinese Journal of Chromatography, 2004, 22(2): 174
丁永胜, 牟世芬. 色谱, 2004, 22(2): 174
[7] Ma P Y, Zhang Y L, Du S. Science and Technology Economic Guide, 2016(6): 121
马骋远, 张宇丽, 杜爽. 科技经济导刊, 2016(6): 121
[8] GB/T 17481-2008
[9] Liu M X, Hu C X, Ren L, et al. Journal of Food Safety and Quality, 2013, 4(6): 1880
刘美霞, 胡彩霞, 任丽, 等. 食品安全质量检测学报, 2013, 4(6): 1880
[10] Lin L, Wang H B. Chinese Journal of Health Laboratory Technology, 2013, 23(7): 1706
林立, 王海波. 中国卫生检验杂志, 2013, 23(7): 1706
[11] Hu J Z, Wei H, Ren H. China Feed, 2003(20): 19
胡京枝, 魏红, 任红. 中国饲料, 2003(20): 19
[12] Zhang H M. Cereal and Feed Industry, 2015(4): 68
张红梅. 粮食与饲料工业, 2015(4): 68[13] Liu Y M, Li G F, Fan X M, et al. Jiangxi Feed, 2002(6): 21
刘耀敏, 李国富, 范小敏, 等. 江西饲料, 2002(6): 21
[14] Yang H Y. Tianjin Agricultural Sciences, 2010, 16(2): 72
杨鸿雁. 天津农业科学, 2010, 16(2): 72
[15] Liu X M, Liu B, Guo Y, et al. Chinese Journal of Food Hygiene, 2016, 28(2): 219
刘雪梅, 刘波, 郭园, 等. 中国食品卫生杂志, 2016, 28(2): 219[16] Wang J J, Zhang W, Tan B Y. et al. Acta Nutrimenta Sinica, 2011, 33(6): 607
王佳佳, 张伟, 谭炳炎, 等. 营养学报, 2011, 33(6): 607[17] Zhang Y F, Zhao S, Wang J. et al. Chinese Journal of Forensic Medicine, 2013, 28(6): 475
张云峰, 赵森, 王炯, 等. 中国法医学杂志, 2013, 28(6): 475
[18] Matuszewski B K, Constanzer M L, Chavez-Eng C M. Anal Chem, 2003, 75: 3019
[19] Tang C L, Wang L, Cheng M C, et al. Chinese Journal of Chromatography, 2015, 33(7): 699
唐春兰, 王莉, 程孟春, 等. 色谱, 2015, 33(7): 699
Determination of choline chloride in milk powder and jelly by solid phase extraction-ultra high performance liquid chromatography-tandem mass spectrometry
WANG Hui*, LIU Jiang, LI Xihui, CAO Yang, HUANG Xiaobei, LI Yongqiang
(ChangshaInstituteforFoodandDrugControl,FoodSafetyProductionEngineeringResearchCenterofHunanProvince,Changsha410013,China)
An analytical method was developed for the determination of choline chloride in milk powder and jelly by solid phase extraction-ultra high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). The samples were centrifuged after hydrolyzation 3 h in 10 mL 0.02 mol/L ammonium acetate (glacial acetic acid adjust pH to 3.0). The supernatant was cleaned up by a DIKMA ProElut PLS column (60 mg/3 mL). The target compounds were separated by an Agilent ZORBAX 300 SCX column (150 mm×2.1 mm, 5 μm). Qualitative and quantitative analyses were carried out by multiple reaction monitoring (MRM) mode with positive electrospray ionization (ESI+). The limit of detection (LOD,S/N=3) and the limit of quantitation (LOQ,S/N=10) were 0.15 mg/kg and 0.50 mg/kg, respectively. The recoveries of choline chloride spiked in milk powder and jelly were 70.8%-100.2%, with RSDs no more than 6.83% (n=6). The standard curves showed good linearity in the concentration range from 0.05 to 8.0 mg/L with correlation coefficient (r) of 0.996. The method was applied to detection of commercially available choline chloride in milk powder and jelly. The contents of choline chloride in real milk power and jelly samples were 251.0-2 448 mg/kg and 0.261-0.314 mg/kg, respectively. The developed method is accurate, reliable, sensitive, and suitable for the determination of choline chloride in milk powder and jelly.
ultra high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS); solid phase extraction (SPE); choline chloride; milk powder; jelly
10.3724/SP.J.1123.2016.12024
2016-12-12
湖南省科技计划重点项目(2014SK2011).
Foundation item: Key Program of Hunan Provincial Science and Technology Department (No. 2014SK2011).
O658
A
1000-8713(2017)05-0558-05
* 通讯联系人. E-mail:wanghuei158@163.com.
猜你喜欢
杂志排行

色谱的其它文章
- 高效液相色谱-串联质谱法测定蜂蜜中20种全氟烷基化合物
- 气相色谱法测定车用汽油中含氧化合物和苯胺类化合物
- 在线固相萃取-离子色谱法测定4种芳环磺酸盐中的硫酸根离子
- 分散固相萃取-液相色谱-串联质谱法测定谷物、蔬菜、水果中27种新型杀菌剂
- Enantioseparation of 2-(substituted phenyl)propanoic acids with hydroxypropyl-β-cyclodextrin as a chiral additive:investigation of substituent influence on enantiorecognition
- 超高效液相色谱/静电场轨道阱高分辨质谱法同时测定塑料食品接触材料中光稳定剂和抗氧化剂的特定迁移量