多位点序列分析揭示我国16SrI组植原体不同株系间遗传变异和系统发育关系*
2017-04-27于少帅任争光宋传生林彩丽朴春根田国忠
于少帅 李 永 任争光 宋传生 林彩丽 朴春根 田国忠
(中国林业科学研究院森林生态环境与保护研究所 国家林业局森林保护学重点实验室 北京 100091)
多位点序列分析揭示我国16SrI组植原体不同株系间遗传变异和系统发育关系*
于少帅 李 永 任争光 宋传生 林彩丽 朴春根 田国忠
(中国林业科学研究院森林生态环境与保护研究所 国家林业局森林保护学重点实验室 北京 100091)
【目的】16SrI组植原体对我国作物和生态植物危害严重。目前植原体尚不能分离培养,对于我国植原体不同株系间遗传变异和种群结构尚不清晰,通过多位点序列分析(MLSA)揭示我国不同地区16SrI组不同植原体株系的遗传多样性及其地理分布特征,比较不同植原体株系间不同持家基因的变异程度,为我国不同植原体株系的检测、分类鉴定和系统发育研究提供一定的参考方法和依据。【方法】应用rp,tuf,secA,secY,ipt,dnaK,fusA,gyrB,pyrG和rpoB共10个持家基因序列,结合16S rDNA序列,以全基因序列已完成的洋葱黄化植原体(OY-M)、翠菊黄化丛枝植原体(AYWB)、澳大利亚葡萄黄化植原体(CPA)、草莓致死黄化植原体(SLY)和苹果簇生植原体(CPM)共5种植原体株系为参照,分析我国10个省(市)苦楝丛枝、莴苣黄化、桑萎缩、泡桐丛枝和长春花绿变植原体株系(共18株)的遗传变异规律和系统发育关系,通过多重序列比对分析不同基因片段的序列多态性和变异程度。【结果】 我国18株苦楝丛枝、莴苣黄化、桑萎缩、泡桐丛枝和长春花绿变植原体株系的rp,tuf,secA,secY,ipt,dnaK,fusA,gyrB,pyrG和rpoB多位点序列共有15种序列类型,从而揭示16SrI组不同植原体株系间丰富的遗传多样性。基于rp等10个基因多位点序列分析可将16SrI-B、D亚组的不同植原体株系清晰地分开。10株苦楝丛枝植原体株系与2株桑萎缩植原体株系系统发育关系最近,多位点序列分析可将这2种基于16S rDNA序列难以区分的植原体株系清晰地区分。10株苦楝丛枝植原体株系分为4个进化枝,其多位点序列存在8种序列类型,这4个分枝与植原体株系的地理分布关系密切。与我国长春花绿变和泡桐丛枝植原体株系相比,福建三明2株莴苣黄化植原体株系与日本洋葱黄化植原体株系OY-M系统发育关系较近。在已检测分析的植原体不同基因序列片段中,dnaK基因序列片段的变异水平最高。【结论】多位点序列分析可做为一种对植原体鉴定、区分以及株系遗传多样性全面检测的有效、可靠方法。在以后的研究中该方法可广泛应用于深入探讨植原体不同组间或亚组间株系的遗传变异和系统发育关系。
植原体; 多位点序列分析(MLSA); 遗传变异; 序列类型(ST); 系统发育
Plant-pathogenic phytoplasmas are phloem-limited prokaryotes lacking cell walls. Diseases associated with these organisms severely damage a wide range of plants (Bertaccinietal., 2009; Leeetal., 2004). Under natural conditions, phytoplasmas are transmitted by insects and grafting. Following infection, the phytoplasmas secrete pathogenic substances, thereby causing diseases with symptoms of witches’-broom, phyllody, leaf yellowing, virescence etc. (Hogenhoutetal., 2008). Because they cannot be maintained in axenic culture, traditional phenotypic characters used for classification of culturable microbes are not suitable for studying phytoplasmas (Dickinsonetal., 2013). Following development of molecular biology, genetic analyses have been used for their detection, identification, and classification. 16S rDNA sequences, which serve as gold standards for determination of bacterial species, has been widely applied to analyzing the phylogenetic relationships of phytoplasmas, followed by sequence analysis of ribosomal protein (rp),tuf,secY, andsecAgene fragments (Hodgettsetal., 2008; Leeetal., 2006;Schneideretal., 1997). The phytoplasmas have been identified to be a large monophyletic group within the mollicutes and to be closely related toAcholeplasmalaidlawii, a member of classMollicutes, primarily based on 16S rRNA andrpgene sequence analyses (Gundersenetal., 1994). At present, 28 groups of phytoplasmas have been classified mainly on the basis of the sequences of 16S rRNA and a few other conserved genes from several hundred phytoplasma strains worldwide (Weietal., 2007). However, sequencing of 16S rDNA—a highly conserved fragment—alone may not be sufficient to guarantee species identity (Foxetal., 1992; Leeetal., 2004). If the phytoplasma strains share >97.5% sequence similarity in their 16S rDNA sequences, they would cluster into the same group and may not be readily differentiated and classified further (Bertaccinietal., 2009; Leeetal., 2004). Since the organisms possess prominently different biological properties and occupy diverse ecological niches (i.e., specific host plants, vectors, or geographical regions) (Bertaccinietal., 2009; Leeetal., 2004), analysis of a limited number of genes would provide insufficient or misleading information for studying phylogenetic relationships at the species and within-species levels (Firraoetal., 2013). To date, sequencing of five complete genomes and six draft genomes of phytoplasmas has been completed, and genetic diversity of phytoplasmas has been explored through comparative analysis of these eleven genomes (Andersenetal., 2013; Baietal., 2006; Kubeetal., 2008; Oshimaetal., 2004; Tran-Nguyenetal., 2008). Whole genome sequencing of phytoplasma genome for studying their biological and ecological properties is relatively difficult and not suitable for genetic analysis of multiples strains.
Multilocus sequence analysis (MLSA) is gradually being recognized as a precise and unambiguous method to investigate the variations in nucleotide sequences and to analyze the phylogenies of microorganisms with close genetic interrelatedness using nucleotide sequence data (Maidenetal., 1998;Saux, 2013). This method was first used in the identification and classification ofNeisseriameningitidess(Maidenetal., 1998). MLSA generates abundant sequence data, which allows discrimination between the large number of isolates obtained in local and global epidemiological investigations (Maiden, 2006;Saux, 2013). For this reason, this approach is being increasingly used in the identification, classification, and phylogenetic analysis of closely related microorganisms (Maiden, 2006). MLSA is particularly popular for the identification of pathogenic isolates associated with different ecological niches (plant hosts, insect vectors, or geographical distributions) (Maiden, 2006). In summary, it allows examination of the phylogenetic relationships, species identification, and discrimination between closely related but definitely distinct strains of phytoplasma (Arnaudetal., 2007; Danetetal., 2007; Lietal., 2014; Musicetal., 2011).
Five economically and environmentally harmful plant diseases associated with AY phytoplasma group (16SrI) have been reported widely in China: chinaberry witches’-broom, lettuce yellows, mulberry dwarf, paulownia witches’-broom and periwinkle virescence, the associated diseases (Lietal., 2005; Tianetal., 1996). Some of these diseases have also been reported in Australia and other Asian countries such as Korea, Japan, India, and Iran. The 16SrI group of phytoplasmas comprises AY phytoplasma (CandidatusPhytoplasma asteris) and numerous related phytoplasmas, infecting a wide range of host plants globally, and represents the most diverse and widespread phytoplasma group (Leeetal., 2004). Within this large and intricate group, the genetic variation and population structure of many closely related phytoplasma strains are still not fully understood and characterized. In China, little is known about the phylogenetic relationships and strain population structure of the five phytoplasmas that are important from the perspective of agriculture and forestry. Furthermore, there is no report on an MLSA specific for this large group of phytoplasma strains, and research on finer identification, genetic structure analysis, and determination of the phylogenetic relationship of the 16SrI group phytoplasmas is limited. Moreover, inadequate knowledge, especially in China, of the relationship between various strains from different plant hosts and locations has greatly retarded detailed phytoplasma identification and differentiation, as well as efficient pathogen detection, disease diagnosis, epidemiological studies, and disease control.
The objectives of the study were: 1) To investigate the phylogenetic relatedness of the phytoplasma strains infecting five kinds of plants—chinaberry (Meliaazedarach), lettuce (Lactucasativa), mulberry (Morusbombycis), paulownia (Paulowniaspp.), and periwinkle (Catharanthusrosea)—from ten provinces (municipality) of China, by using sequence analysis of the 16S rDNA and 10 house keeping genes, they arerp,tuf,secA,secY,ipt,dnaK,fusA,gyrB,pyrG, andrpoBgenes and the concatenated sequence dataset of the ten gene fragments. 2) To elucidate the host specificity and genetic diversity of each strain from the 16SrI phytoplasma group and its relationship with geographical distribution. 3) To compare the discriminatory power of the distinct genes used in this study with those in the previous investigation.
1 Materials and methods
1.1 Isolates and nucleic acid preparation
16SrI group phytoplasma strains used in the study were collected from different regions of China to represent diverse environmental niches of these organisms. These strains were composed of five kinds of phytoplasmas, namely, chinaberry witches’-broom phytoplasma (CWB; from Sanya in Hainan, Fuzhou, Fuqing, and Yong’an in Fujian, Nanjing in Jiangsu, Nanchang in Jiangxi, Changsha in Hunan, and Guangzhou in Guangdong), lettuce yellows phytoplasma (LY; from Sanming in Fujian), mulberry dwarf phytoplasma (MD; from Hefei in Anhui and Chun’an in Zhejiang), paulownia witches’-broom phytoplasma (PaWB; from Yanzhou in Shandong, Nanjing in Jiangsu, and Huairou in Beijing), and periwinkle virescence phytoplasma (PeV; from Haikou in Hainan). The PaWB strain from Yanzhou, Shandong was maintained as a paulownia tissue-culture seedling in the tissue culture chamber of the Research Institute of Forest Ecology, Environment and Protection, Chinese Academy of Forestry. Plant materials infected by representative phytoplasma strains were collected from ten Chinese provinces (municipality) during 2013—2014. Total DNA was extracted from leaf midribs or other tissues of these plants by using a DNA extraction kit (CTAB Plant Genome DNA Rapid Extraction Kit, Aidlab Biotechnologies Co., Ltd, Beijing). DNA and related plant materials were stored in the Research Institute of Forest Ecology, Environment and Protection, Chinese Academy of Forestry.
Two 16SrI strains (onion yellows phytoplasma (OY-M) and aster yellows witches’-broom phytoplasma (AYWB)), one 16SrX strain (CandidatusPhytoplasma mali AT (CPM)), and two 16SrXII strains (CandidatusPhytoplasma australiense (CPA) and strawberry lethal yellows phytoplasma (SLY)), their whole genomes have been sequenced, were employed as references and outgroups (Andersenetal., 2013; Baietal., 2006; Kubeetal., 2008; Oshimaetal., 2004; Tran-Nguyenetal., 2008). These genome sequences and other DNA sequences used in the 16S rDNA analysis were obtained from National Center for Biotechnology Information (U.S. National Library of Medicine, Bethesda, MD, U.S.A.). Further information of all phytoplasma strains employed in the research is listed in Tab.1.
1.2 Genes selected and primers designed
16S rDNA was amplified using primer pairs P1/P7 (Dickinsonetal., 2013). Other studied genes were as follows:rp, including fragments ofrpl22 andrps3 (encoding ribosomal proteins);tuf(elongation factor Tu);secA(preprotein translocase subunit SecA);secY(preprotein translocase subunit SecY);ipt(tRNA delta(2)-isopentenyl-pyrophosphate transferase);dnaK(molecular chaperone DnaK);fusA(elongation factor G);gyrB(DNA gyrase subunit beta);pyrG(CTP synthase); andrpoB(DNA-directed RNA polymerase subunit beta). Primer pairs fordnaK,fusA,gyrB,pyrG, andrpoBwere designed and synthesized based on the genomic sequence of the OY-M isolate (AP006628) by using the Primer Premier 5.0 software (PREMIER Biosoft International, Palo Alto, CA, U.S.A.), while those forrp,tuf,secA,secY, andiptwere selected from previous studies (Hodgettsetal., 2008; Huetal., 2013; Leeetal., 2003;Schneideretal., 1997). All primer sequences are shown in Tab.2.
1.3 PCR amplification and DNA sequencing
The PCR mixture (25 μL) contained 1 μL undiluted DNA preparation, 0.5 μL forward/reverse primer (10 μmol·L-1), 12.5 μL 2× PCR Master Mix (including 0.05 U·μL-1Taq DNA polymerase, 4 mmol·L-1MgCl2, 0.4 mmol·L-1dNTPs), and ddH2O and was purchased from Biomed (Beijing). PCR conditions are detailed in Tab.2. DNA from five types of healthy host plant materials was used as the negative control. Amplicons were analyzed by electrophoresis in 1% (W/V) agarose gel, stained using SYBR Green I, and visualized with a gel imaging system. Partial nucleotide sequences of the 11 studied genes were obtained by direct sequencing of the PCR products, which were purified and sequenced on an ABI PRISM 3700 automated DNA sequencer by a service provider (Sangon, Beijing).
1.4 Sequence analysis
The five kinds of experimental phytoplasma strains were identified and classified using restriction fragment length polymorphism (RFLP) analysis with an interactive online tool for phytoplasma classification,iPhyClassifier, based on their 16S rRNA gene sequences (Weietal., 2007; Zhaoetal., 2009). All sequences used in this study were assembled using DNAMAN version 5.0 (Lynnon Corporation, Vaudreuil-Dorion, Quebec, Canada) and were edited using EditSeq (DNAStar package, Madison, WI, USA). All sequences were aligned by multiple sequence alignment using DNAMAN version 5.0. Phylogenetic analyses were performed by adopting neighbor-joining (NJ) method using the molecular evolutionary genetics analysis (MEGA) software version 4.0. Bootstrap analyses were performed with 1 000 replicates. The complete sequence of each analyzed gene was selected from the OY-M genome and used as a putative consensus sequence. The original nucleotide positions of the consensus fragments in the OY-M complete genome were 279425-280928 (16S rDNA), 245976-247089 (rp), 305131-306315 (tuf), 530192-532699 (secA), 252238-253479 (secY), 316651-317526 (ipt), 795359-797224 (dnaK), 302934-305000 (fusA), 562356-564371 (gyrB), 689018-690640 (pyrG), and 294025-297846 (rpoB). The polymorphic site positions of the analyzed genes were numbered corresponding to the putative consensus gene sequence. Allele numbers were assigned to each unique sequence. Each isolate was defined by an allele profile or sequence type based on the combination of numbers corresponding to alleles at the loci analyzed. The percentage of polymorphic sites in amplified sequences was calculated to evaluate variations in each gene. Parameters, including length amplified (bp), number of alleles and polymorphic sites, percentage of variable loci in the analyzed sequence (%), percentage of transition, transversion, deletion, or insertion in the variable loci (%), that assessed the variable status of gene fragments of phytoplasmas were computed (Fraseretal., 2004).
1.5 Nucleotide sequence accession numbers
The nucleotide sequences reported in the study have been deposited in the GenBank database, and the accession numbers are listed in Tab.1.

Tab.2 Universal and specific primers used to amplify 16S rDNA, rp, tuf, secA,secY, ipt, dnaK, fusA, gyrB, pyrG and rpoB gene fragments①
① Amplification parameters: 94 ℃ for 5 min, 35 cycles of 94 ℃ for 30 s, 53 ℃ for 40 s and 72 ℃ for 2 min (a); 35 cycles of 94 ℃ for 30 s, 52 ℃ for 40 s and 72 ℃ for 1 min 20 s (b); 35 cycles of 94 ℃ for 30 s, 53 ℃ for 40 s and 72 ℃ for 1 min (c); 35 cycles of 94 ℃ for 30 s, 55 ℃ for 40 s and 72 ℃ for 1 min 30 s (d); 35 cycles of 94 ℃ for 30 s, 55 ℃ for 40 s and 72 ℃ for 1 min (e); 35 cycles of 94 ℃ for 30 s, 55 ℃ for 40 s and 72 ℃ for 1 min 20 s (f); 35 cycles of 94 ℃ for 30 s, 52 ℃ for 40 s and 72 ℃ for 1 min 30 s (g); 35 cycles of 94 ℃ for 30 s, 53 ℃ for 40 s and 72 ℃ for 1 min 20 s (h), followed by 72 ℃ for 10 min.
2 Result
2.1 16S rRNA gene sequence analysis
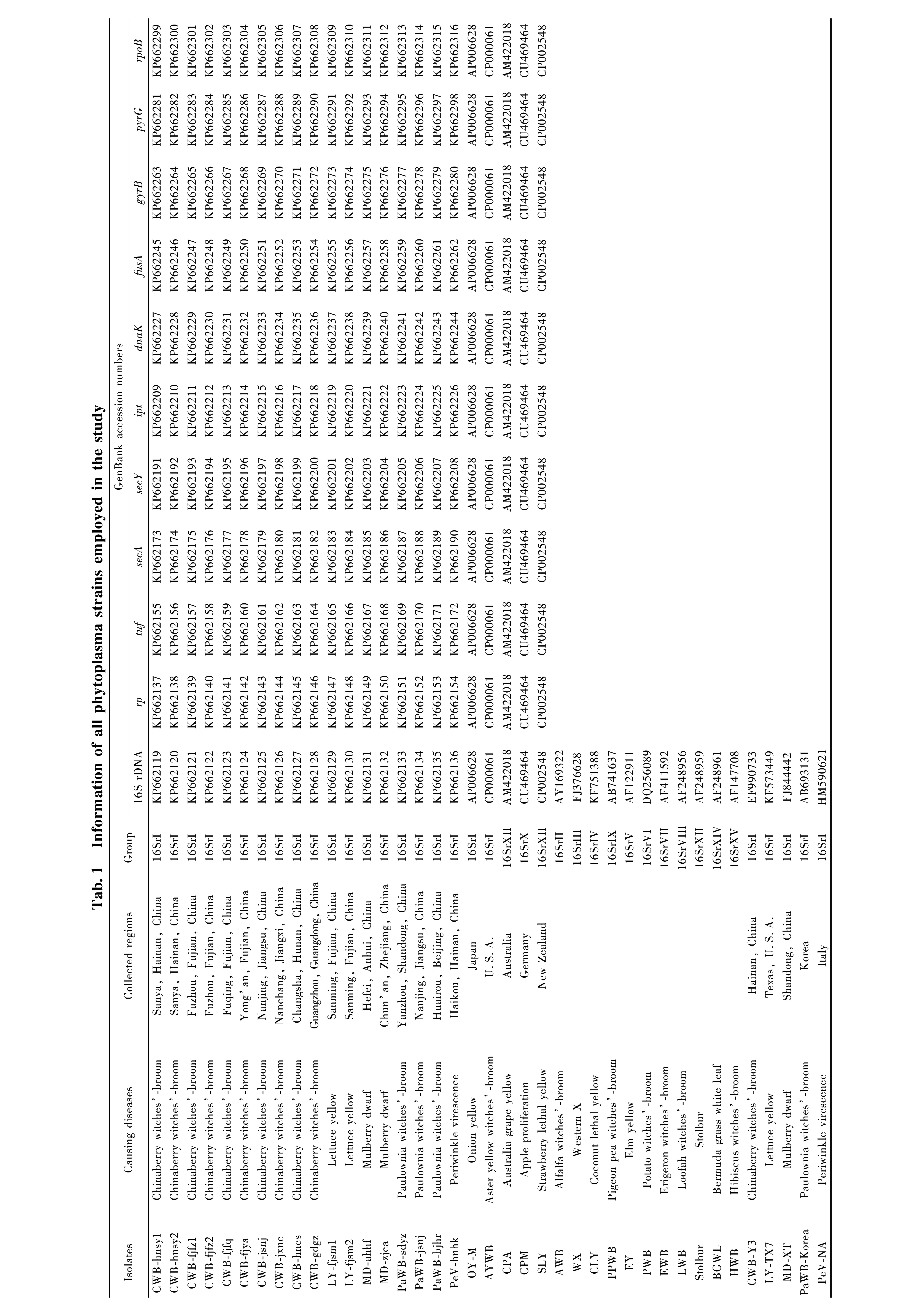
The 16S rRNA gene sequences of the studied phytoplasma strains were identical to those of reference strains of the 16SrI phytoplasma group obtained from theiPhyClassifier database (Zhaoetal., 2009). Based on the RFLP analysis of 16S rRNA gene sequences, some tested phytoplasma strains of the 16SrI group from diverse ecological conditions, such as CWB, MD, and PeV strains, could not be differentiated. The five phytoplasma strains appeared to be highly homogeneous based on 16S rDNA sequences, sharing 99.6%-100% sequence identity. The phylogeny analysis indicated that CWB, LY, MD, and PeV strains from different regions of China, the reference OY-M strain of 16SrI-B subgroup from Japan (Leeetal., 2004), the PaWB strains of 16SrI-D subgroup from China and Korea (Leeetal., 2004), and the reference AYWB strain of 16SrI-A subgroup from North America formed a discrete clade with a bootstrap support value of 99%. Three PaWB strains from Beijing, Shandong, and Jiangsu and a Korean strain were clustered with a 67% bootstrap value, while two LY strains from Fujian were clustered with a 96% bootstrap value. However, the 16S rDNA sequences of 10 CWB strains, including the reference strain CWB-Y3 from Hainan, and 3 MD strains, including the MD-XT reference strain from Shandong and lettuce yellows LY-TX7 reference strain from Texas, U.S.A., were highly similar and hardly differentiated (Fig.1).

Fig.1 Phylogenetic tree constructed by neighbor-joining analysis of 16S rRNA gene sequences fragments from phytoplasma strains Bar length represents inferred character-state changes. Branch lengths are proportional to the number of inferred character-state transformations. Bootstrap values (1 000 replicates) are shown on branches.The same below.
2.2 Comparative analysis of housekeeping gene fragments
Based on 10 fragments of housekeeping genes obtained in the study, all 18 phytoplasma isolates from China shared sequence identity of 99.5%-100% inrp, 99.9%-100% intuf, 99.0%-100% insecA, 98.8%-100% insecYandfusA, 98.9%-100% iniptanddnaK, 99.1%-100% ingyrB, 98.4%-100% inpyrG, and 98.6%-100% inrpoB. Phylogenetic analysis of a single locus showed that the AYWB and OY-M from Japan belonging to subgroup B could be clearly distinguished from all 10 genes from five kinds of 16SrI group strains from China, suggesting that 16SrI-B subgroup is distantly related to the 16SrI-A subgroup. In addition, PaWB and LY could also be distinguished from other strains based on theirrp,tuf, andsecAsequences, but the differentiation of CWB and MD strains was ambiguous. However, approximately five kinds of strains infecting different plants, including CWB and MD, were differentiated clearly based on theirsecY,ipt,dnaK,fusA,gyrB,pyrG, andrpoBsequences, with relatively high bootstrap support values. Noticeably,secYwas better compared to other housekeeping genes in resolving the host specificity of investigated phytoplasmas.
Nevertheless, based on efficient gene sequences, finer strain lineages could not be delineated in the same kind or different kinds of strains. For instance, phylogenetic analysis based on each gene sequence hardly unraveled the relationship between genetic diversity and geographical distribution among CWB strains from ten provinces (municipality) of China, except for CWB strains from Hainan, which formed an independent subclade during analysis of the gene fragments ofipt,dnaK,fusA,pyrG, andrpoB. LY strains (LY-fjsm1, LY-fjsm2) from Yong’an, Fujian, where widely cultivated lettuce was seriously infested by LY phytoplasma, was most closely related to OY-M or PeV isolates than to the initially suspected local CWB-fjya strains collected from a diseased chinaberry tree at an adjacent site to LY-affected land. As LY and OY-M could not be differentiated on the basis ofgyrBandpyrGsequences, we assumed that these are the same pathogens that infect herbaceous lettuce and onion, respectively.
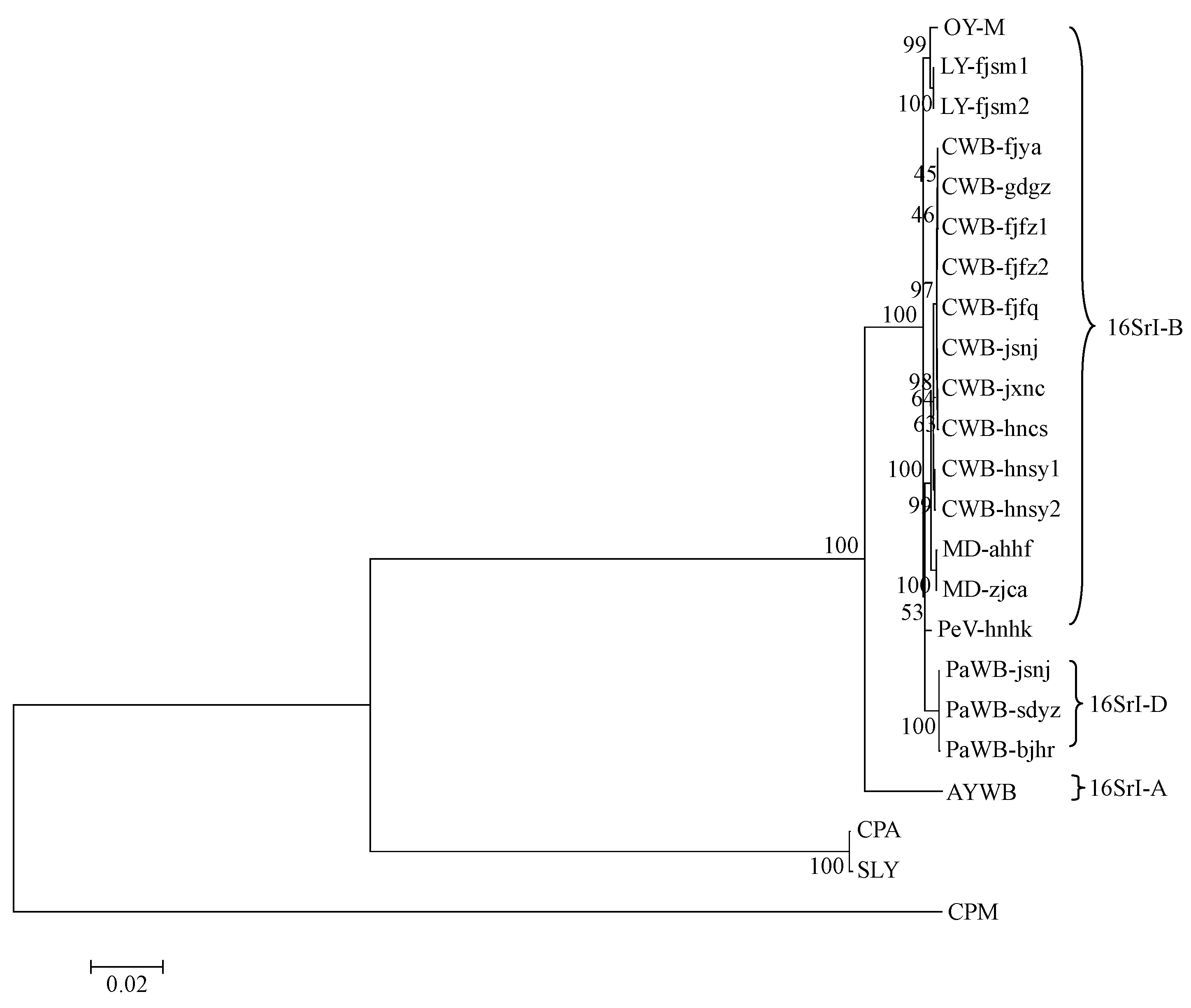
Fig.2 Phylogenetic tree of 16SrI group and other group phytoplasmas constructed by neighbor-joining analysis of the concatenated gene sequences data set of rp, tuf, secA, secY, ipt, dnaK, fusA, gyrB, pyrG and rpoB gene sequences
2.3 MLSA
The concatenated sequence data set (8 939-9 010 bp) of the 10 gene fragments ofrp,tuf,secA,secY,ipt,dnaK,fusA,gyrB,pyrG, andrpoBand corresponding nine putative amino acid sequences (excluding RP) were used for phylogenetic analysis. The 18 16SrI group strains formed a distinct cluster with a 100% bootstrap value, sharing 99.1%-100% sequence identity and showing distant relation to other 16Sr phytoplasmas including CPA, SLY and CPM. This clade also comprised reference strains OY-M and AYWB and was divided into several subclades (Fig.2). To further estimate interrelatedness of the 16SrI group phytoplasma strains, the phylogenetic tree composed of only the 16SrI group strains was reconstructed based on the concatenated gene sequence data set of the 10 loci (Fig.3). Based on MLSA, significant and distinct differentiations were observed that not only correlated with the specificity of plant hosts but also with geographic origins of the analyzed isolates, which were hardly distinguished by the RFLP analysis of their 16S rRNA gene sequences. The CWB, LY, MD, PaWB, PeV, OY-M, and AYWB strains were clearly differentiated and formed several discrete subclades with improved meaningful bootstrap support values. Significantly, 10 CWB isolates from diverse regions were further classified into four subclusters, namely CWB cluster 1 (one strain from Jiangsu), CWB cluster 2 (two strains from Jiangxi and Hunan), CWB cluster 3 (including five strains from Fujian and Guangzhou), and CWB cluster 4 (two strains from Hainan). An explanatory chart was drawn to clarify the close relationships between the phytoplasma strains of different CWB genotypes and their geographical distributions (Fig.4), which were hardly dissected by previous single locus studies (Lietal., 2005). The MLSA results of concatenated sequences of nine proteins were in agreement with those of nucleotide sequences, despite some differences in the branching topological structure.
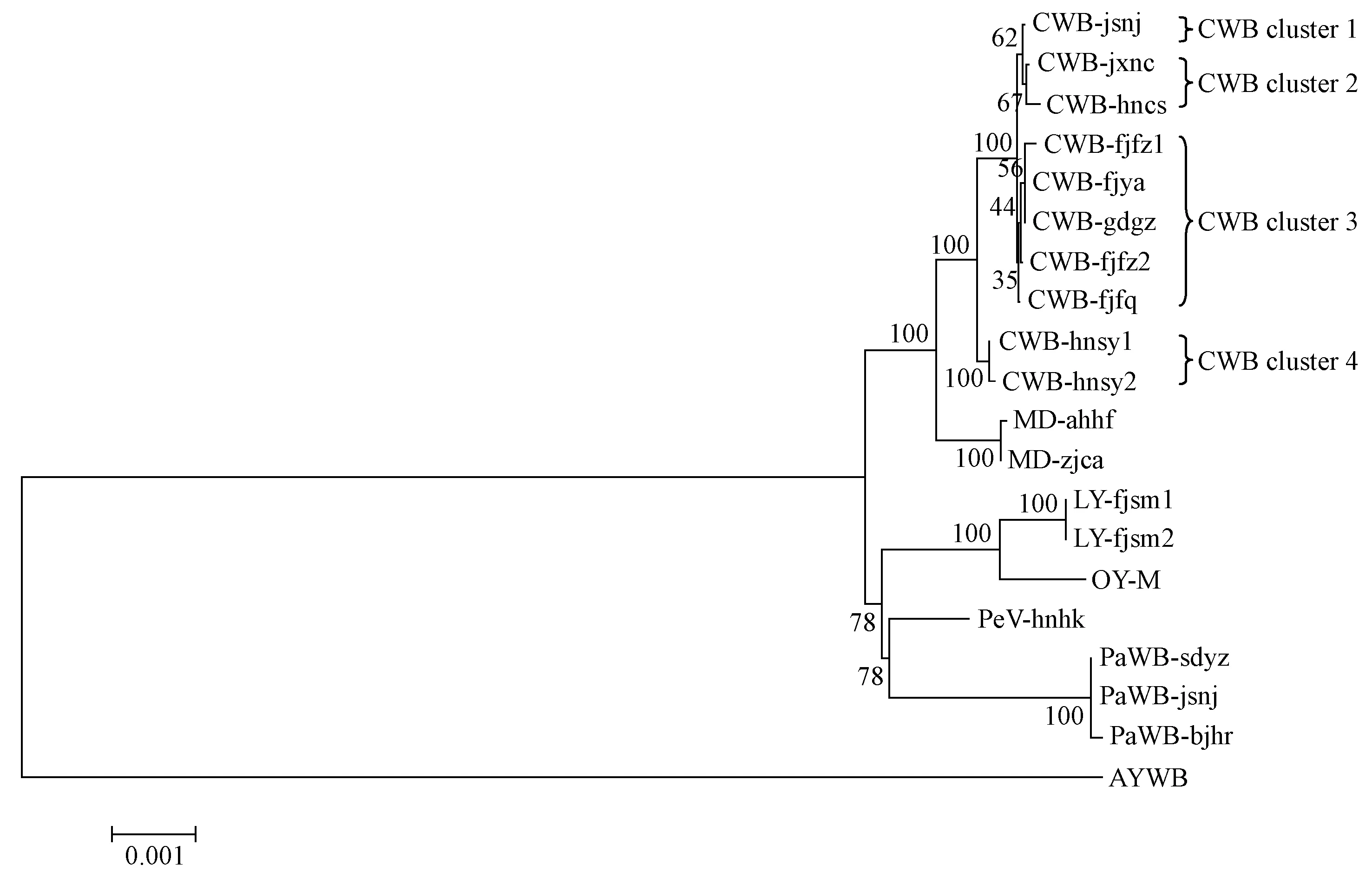
Fig.3 Phylogenetic tree of the 16SrI group phytoplasma strains reconstructed by neighbor-joining analysis of the concatenated gene sequences data set of rp, tuf, secA, secY, ipt, dnaK, fusA, gyrB, pyrG and rpoB gene sequences
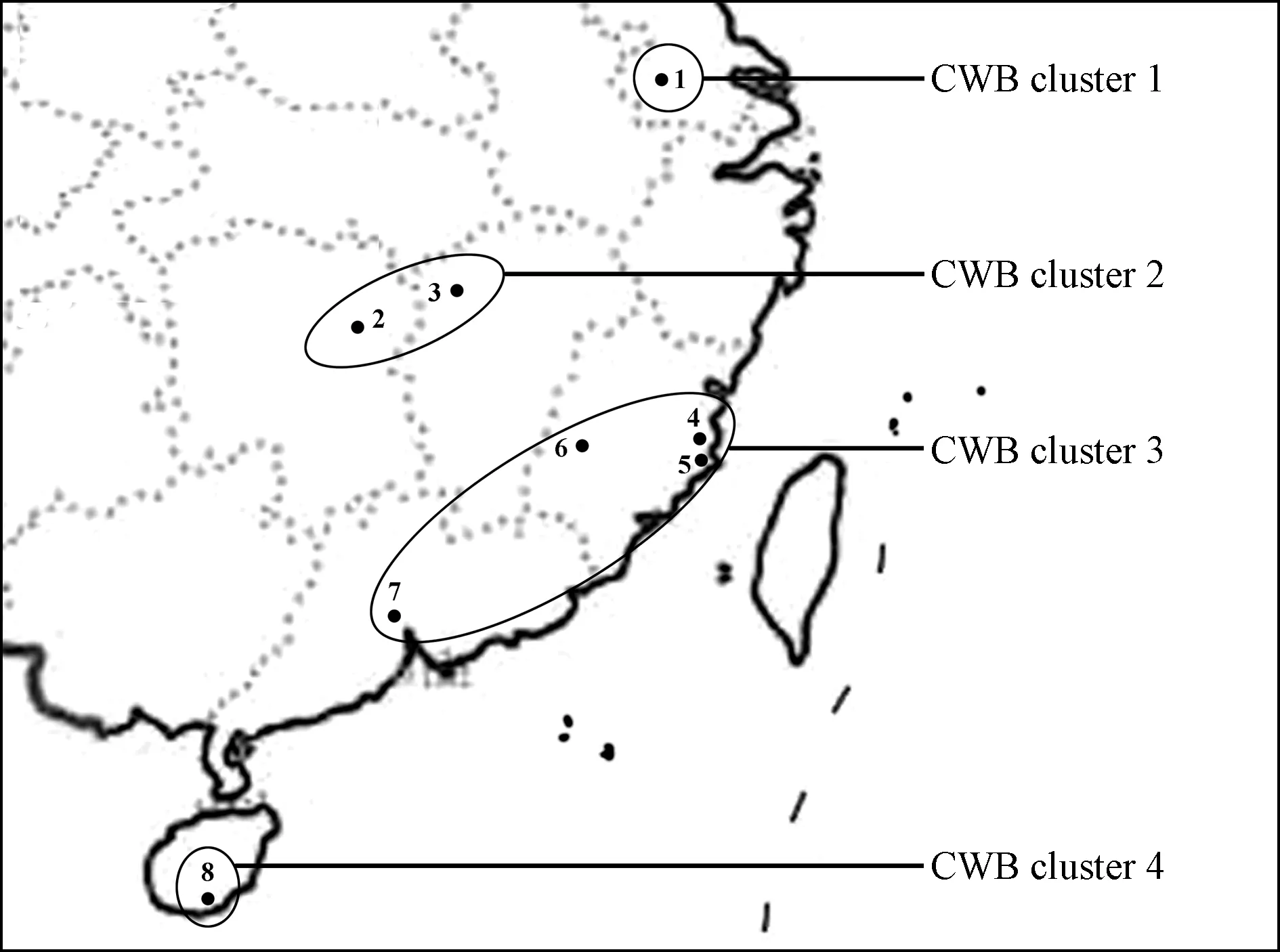
Fig.4 Geographical locations of the four CWB cluster strains in China The background geographical map was partial map of China. 1: Nanjing, Jiangsu; 2: Changsha, Hunan; 3: Nanchang, Jiangxi; 4: Fuzhou, Fujian; 5: Fuqing, Fujian; 6: Yong’an, Fujian; 7: Guangzhou, Guangdong; 8: Sanya, Hainan.
2.4 Polymorphic sites and DNA sequence types

Fig.5 Polymorphic nucleotide sites in 16S rRNA, rp, tuf, secA, secY, ipt, dnaK, fusA, gyrB, pyrG and rpoB genes of the 16SrI group phytoplasma strains The complete sequence of each analyzed gene was elected from OY-M (AP006628) complete genome and performed as a putative consensus sequences; only those that differ from the nucleotide in the consensus sequence were displayed for the alleles. A dash (-) denotes a single-nucleotide deletion. Nucleotide positions are numbered in vertical format according to the first nucleotide position of the corresponding gene sequence. The number of the strains possessing the allele is indicated in parentheses.
Polymorphic nucleotides of the 16S rRNA gene,rp,tuf,secA,secY,ipt,dnaK,fusA,gyrB,pyrG, andrpoBwere obtained from the multiple sequence alignment data (Fig.5). The number of alleles in each gene ranged from five (16S rDNA) to nine (dnaK), while the number of polymorphic loci ranged from six (16S rDNA) to 39 (dnaK). Allele frequencies indicated that most loci had one dominant allele in the population, as listed in Fig.5 and Tab.3. Parameters elucidating the variable status of different genes were calculated, as summarized in Tab.4. The variation was comparatively high in sequence fragments ofdnaK(3.35%-3.44%),secY(2.38%-2.47%), andipt(2.05%-2.17%). In contrast, sequences of 16S rDNA (0.42%) andtuf(0.73%-0.74%) were more conserved, as indicated by lower variation in these genes. There were 201 variable loci in the 11 analyzed gene sequences. The level of transition in mutant loci (57.71%) was definitely higher than that of transversion in mutant loci (36.82%). With regard to nucleotide substitution in 201 variable loci, transition between T and C (33.83%) was more frequent compared with that between A and G (23.88%). Transversion between A and C (17.41%) and T and G (12.44%) was common, while transversion between G and C (2.99%) and A and T (3.98%) was comparatively rare. Furthermore, deletion and insertion (6.47%) existed inrp,tuf,secY, anddnaKsequences. The deletion and insertion of nucleotides in the objective gene resulted in the addition or absence of corresponding amino acids in the deduced amino acid sequence encoded by the gene. Based on the analysis of the 11 gene fragments, there were 15 sequence types (STs) in 18 strains (Tab.3). Ten CWB strains were resolved into 8 STs, while three strains, CWB-fjfz2, -fjya, and -gdgz had same ST although they had different geographical origins (Fuzhou and Yong’an in Fujian and Guangzhou in Guangdong, respectively). Both LY strains from Sanming, Fujian (LY-fjsm1, -fjsm2) shared the same ST.
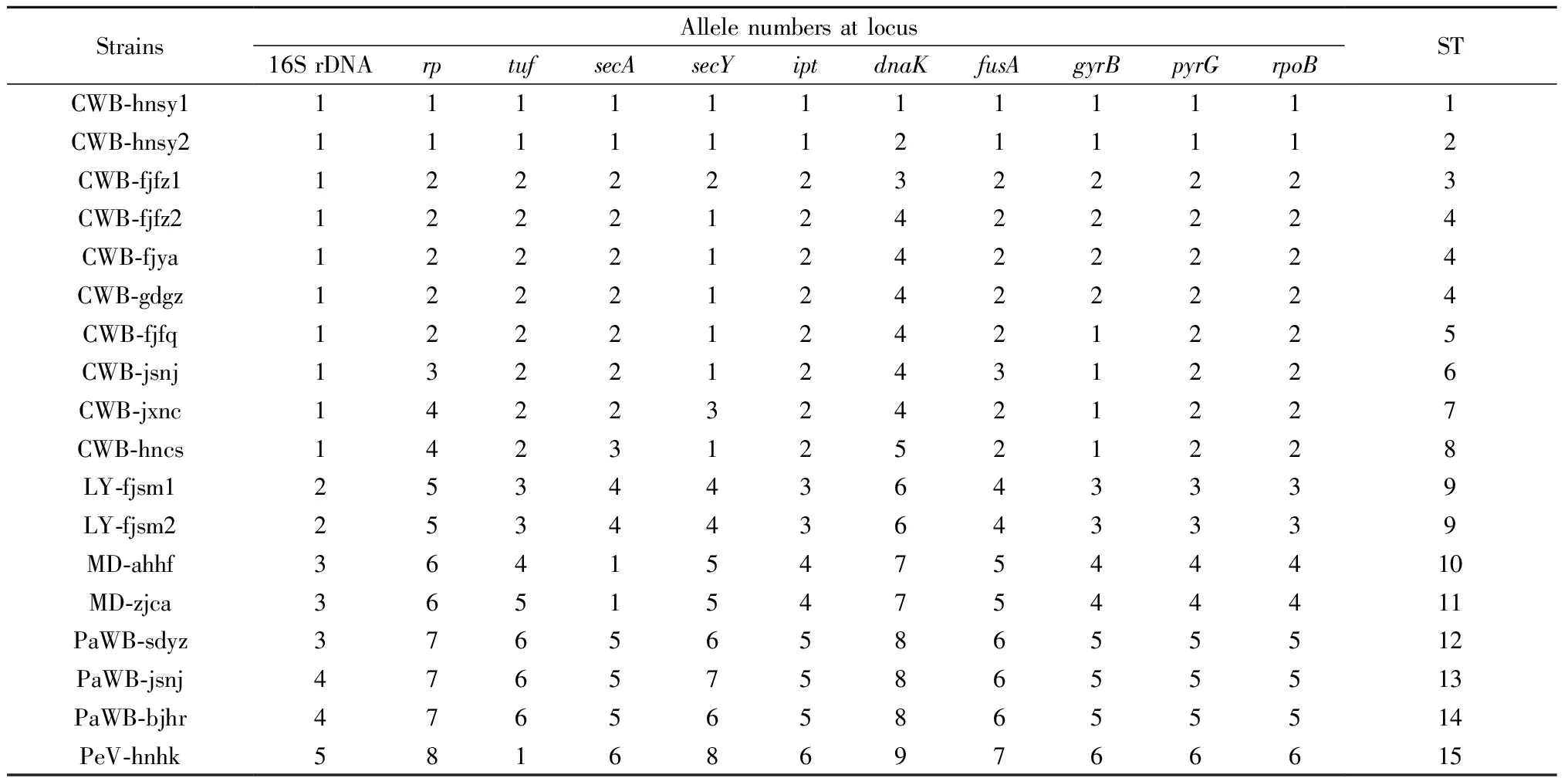
Tab.3 DNA sequence types of the 16SrI group phytoplasma strains①
① Arabic numbers in the table are allele numbers of each unique sequence as described in ‘1.4’. ST represents sequence type.
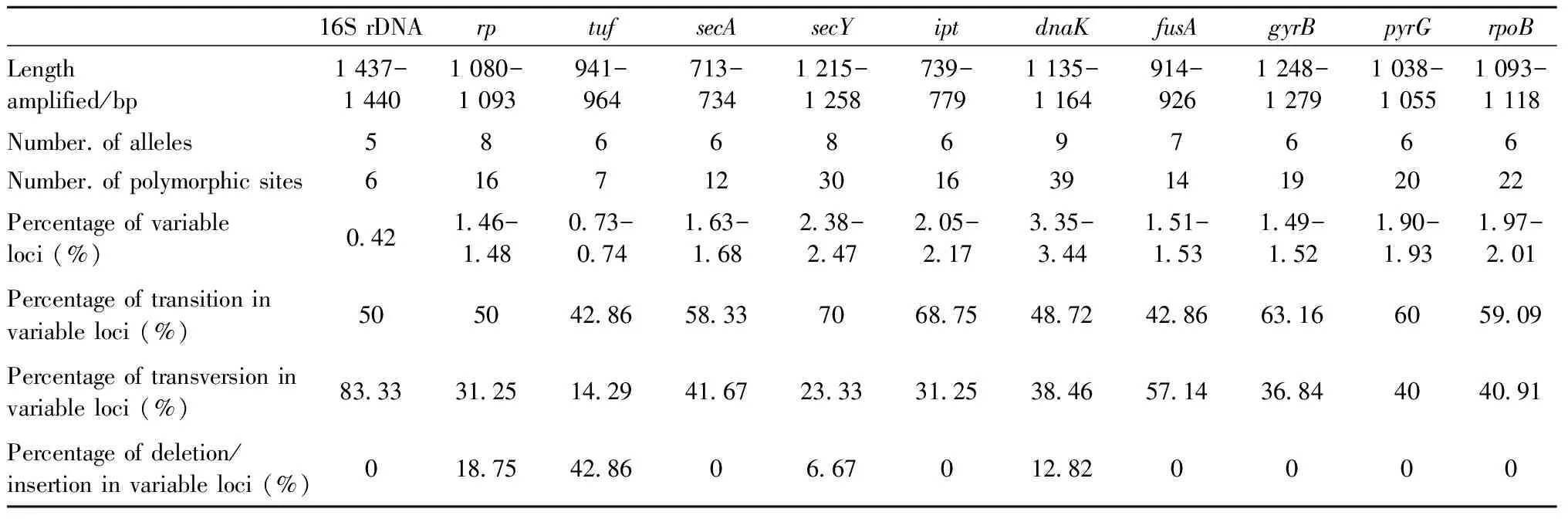
Tab.4 Variation status of the eleven gene sequence fragments in the phytoplasmas associated with five important plant diseases from different locations of China
3 Discussion
Arnaudetal. (2007) confirmed the close genetic interrelatedness of three distinct Flavescence Dorée phytoplasma strain clusters and FD-related phytoplasmas belonging to the 16SrV group infecting grapevine, alder, elm, blackberry, and Spanish broom in Europe by MLSA of themap,uvrB-degV, andsecYgenes. This finding suggested that phytoplasmas causing FD, German Palatinate grapevine yellows, and alder yellows might have originated in Europe. The discrimination of some 16SrX strains ofCandidatusPhytoplasma prunorum,Ca. P. mali, andCa. P. pyri from Azerbaijian, Austria, Croatia, France etc. was first achieved by MLSA of theaceF,pnp,imp, andsecYgenes (Danetetal., 2007). Further, the genetic variability of ten selected 16SrXII-A subgroup stolbur strains from grapevine was investigated by MLSA oftuf,secY, andvmp1 genes (Musicetal., 2011). In China, Lietal. (2014) elucidated the genetic relationships among four phytoplasmas of the peanut witches’-broom group (16SrII-A) by MLSA ofrplV-rpsC,rpoB,gyrB,dnaK,dnaJ,recA, andsecYgenes. Their findings suggested thatCrotalariapallida,Tephrosiapurpurea, andCleomeviscosemay be natural hosts of peanut witches’-broom phytoplasma in the Hainan province. To the best of our knowledge, this is the first study to conduct MLSA of as many as ten housekeeping genes to evaluate the genetic variation and phylogeny of phytoplasma strains of the 16SrI group from diverse environments of China. We successfully identified several novel loci to cover more regions of the phytoplasma chromosome, and this would expand the genome-scanning scope and increase the variation-detection capacity of the technique. Our results allowed for clear differentiation of closely related phytoplasma strains. This improved MLSA method that uses the genes that exist in a single copy as well as more variable genes such asipt,dnaK,fusA,gyrB,pyrG, andrpoBas genetic markers can not only identify and differentiate unambiguously different strains infecting various plant hosts, but also strains from different geographical locations. The strains CWB, LY, MD, PaWB, and PeV and the reference strains OY-M and AYWB were clearly differentiated into seven discrete subclades within the 16SrI group clade. Of note, AYWB, which mainly infects plants in North America and has been classified to the 16SrI-A subgroup, was always clearly distinguished from the strains of the 16Sr-B and 16SrI-D subgroups that originate in Asian regions (Leeetal., 2004), demonstrating apparent ecological separation and divergent evolutionary origins as revealed in 16SrV group phytoplasmas (Laietal., 2014). Unlike AYWB, PaWB strains were closely related to the strains of CWB, LY, MD, PeV and OY-M, while the CWB strains from diverse regions of China were found to be the closest relatives of the MD strains.
Chinaberry witches’-broom disease, which is widespread in southern China, has incidence as high as 50% and causes significant damage to the popular tree chinaberry in urban and rural regions, and the typical symptoms include reduced leaf size, leaf yellowing, shoot proliferation, and progressive decline (Jinetal., 1982). There was no definite difference among the symptoms of chinaberry witches’-broom disease that could be directly used for practicable strain differentiation in the diverse sampling regions in China due to the difficulty ofinvitroculture of phytoplasmas and that the symptom expression and development was greatly influenced by the sampling period as well as sites of different climate zones. From that, we used MLSA to differentiate and determine the close genetic interrelatedness among the causative agent at the molecular level. Intriguingly, ten CWB strains from the Fujian, Guangdong, Jiangsu, Jiangxi, Hunan, and Hainan provinces of south China—a broad scope—showed high diversity and were clearly divided into four clusters or eight putative sequence types. This diversity appeared to be consistent with the difference in the latitude and climate of their specific geographical regions, most possibly attributable to the effect of temperature. Temperature may be the main factor influencing the ecological difference in the diverse natural environments of the CWB strain sampling sites that were distributed across different latitudes from north to south. The CWB strains from Sanya, Hainan province (island), which has a typically tropical climate, showed obvious differences in the sequence ofipt,pyrG,rpoB,tuf,secA,dnaK,fusA,andgyrBgenes from other CWB strains isolated from mainland China. This finding strongly suggested that geographic separation, low latitude, and constantly higher temperature likely acted as selective pressures and influenced the adaptation and evolution of these cell wall-less prokaryotes. Therefore, the notable divergence between the two CWB strains is possibly caused by geographical isolation of the Hainan province from the mainland, which facilitated population differentiation (Fraseretal., 2004). This shows that MLSA provides accurate identification and classification of new variants and confirmation of divergent isolates, which may be of great value for diagnosis, quarantine, epidemiological surveillance, and management of diseases (Maiden, 2006). Our results clarified the systematic classification status of the tested phytoplasma strains from China, and they were generally consistent with the findings of previous investigations (Leeetal., 2004; Tianetal., 1996). However, our findings contrasted those of some previous studies. Phytoplasma strains infecting chinaberry (China tree) were identified as members of the 16SrIII group in Argentina (Galdeanoetal., 2004). A periwinkle yellows phytoplasma strain from Danzhou, Hainan province, was classified in the 16SrV group rather than the 16SrI group (Cheetal., 2009). Lastly, a 16SrV group phytoplasma strain was found to be associated with lettuce yellows disease in the Xinjiang autonomous region of China (Shietal., 2013).
By analyzing nucleotide polymorphisms in eleven genes in the 16SrI group phytoplasma strains, we found that the levels of variations are different in these gene fragments, ranging from high to low in the order ofdnaK,secY,ipt,rpoB,pyrG,secA,fusA,gyrB,rp,tuf, and 16S rDNA. The 16S rDNA sequence, a traditional molecular tool for bacterial taxonomy, is highly conserved and contains comparatively less polymorphic loci (Foxetal., 1992). In comparison, thednaK,secY, andiptgenes possess sufficient polymorphic nucleotide sites to amplify the resolving power such that phylogenetic relationships among diverse phytoplasma strains become apparent. The expression of these genes appears to be associated with the environmental niches of the phytoplasmas to some extent. The proteins encoded bydnaK,secY, andiptplay important roles in the essential metabolism of phytoplasma strains and their interaction with the host plants, insect vectors, or ecological environments (Oshimaetal., 2004). The strains investigated in this work were collected from different plants and diverse ecological conditions. Genetic variation probably occurs in the genomes of these strains in the course of long-term phylogenetic evolution through recombination as well as stabilizing selection, neutral mutation, or positive selection (Fraseretal., 2004). This could be the reason for the relatively high nucleotide polymorphisms in thednaK,secY, andiptgenes. The high variability in thednaKgene likely reflects phytoplasmal adaptation and evolution in response to stress and extreme environments by encoding diverse molecular chaperones to promote cell survival. Therefore, these variations probably play an important role in the differentiation of phytoplasma strains. In contrast to the phytoplasma strains of different subgroups defined by the 16S rRNA gene sequences, the isolates of different subgroups distinguished by the concatenated gene from the ten loci presented a strong association with the specific host plants or geographical locations. This finding likely reflects the differences in the capacity for growth and propagation, pathogenicity, and adaption of the various phytoplasma strains infecting the same or different hosts. Therefore, these genes could be used in combination as a useful tool for precise and unambiguous characterization of phytoplasmas at the strain level, as well as for the strains of other microorganisms that show close genetic interrelatedness.
2.2.3 治疗反应的分析和评估:临床上最初的诊断是拟诊,通常是通过病情的演变和观察治疗反应来明确诊断的。也就是说,如果治疗有效,那么拟诊就可以确立诊断了。例如浆液性结核性胸膜炎胸水中往往找不到结核杆菌,如果临床表现和胸水化验符合结核性胸膜炎,抗结核治疗有效,就可以做出临床诊断了。可见治疗反应的评估与诊断的关系极大。为了鉴别诊断,还需要决定治疗的先后顺序,如1例右上肺炎性阴影合并空洞的患者,肺结核或肺脓肿的可能性都有,先按肺脓肿给予抗生素治疗,结果炎症很快吸收,空洞也关闭。如果先按肺结核治疗,观察的时间就要长得多。诊断就会延迟。
4 Conclusion
The 16SrI group phytoplasma strains analyzed in this study could be distinguished clearly by the improved MLSA method using novel and well-selected marker genes possessing different levels of variability, and the genetic variation and phylogenetic interrelatedness among the isolates closely associated with different plant hosts and geographic distribution were also elucidated unambiguously. Thus, this improved MLSA method allows for precise and consistent molecular typing, identification, and classification of phytoplasmas occupying different ecological niches, except when using 16S rDNA and a few other conserved genes. Moreover, our findings show the analysis of genes with comparatively high numbers of single nucleotide polymorphisms, such asdnaK,secY, andipt, could supplement sequence analysis of 16S rRNA and other conserved genes for genetic analyses as well as accurate detection and identification of these yet-uncultured microorganisms. Meanwhile, several molecular signatures due to the insertion or deletion of nucleotides in certain genes detected in some strains might greatly facilitate the understanding of the relationship of phytoplasmal growth, propagation, and pathogenicity with gene variation and evolution, as well as facilitating the development of novel specific pathogen detection methods. Thus, we expect that the findings from this study, as well as the improved MLSA method for the molecular typing of closely related phytoplasma strains, will promote further detailed investigations into various aspects of phytoplasmas, such as in-depth examination of strain diversity, population structure, and evolution of various 16Sr groups or subgroups; epidemiological studies; and development of reliable pathogen-detection and diagnosis methods.
Reference
Andersen M T, Liefting L W, Havukkala I,etal. 2013. Comparison of the complete genome sequence of two closely related isolates of ‘Candidatusphytoplasma australiense’ reveals genome plasticity. BMC Genomics, 14(1): DOI:10.1186/1471-2164-14-529.
Arnaud G, Malembic-Maher S, Salar P,etal. 2007. Multilocus sequence typing confirms the close genetic interrelatedness of three distinct Flavescence Dorée phytoplasma strain clusters and group 16SrV phytoplasmas infecting grapevine and alder in Europe. Appl Environ Microbiol, 73(12): 4001-4010.
Bai X D, Zhang J H, Ewing A,etal. 2006. Living with genome instability: the adaptation of phytoplasma to diverse environments of their insect and plant hosts. J Bacteriol, 188(10): 3682-3696.
Bertaccini A, Duduk B. 2009. Phytoplasma and phytoplasma diseases: a review of recent research. Phytopathol Mediterr, 48(3): 355-378.
Che H Y, Luo D Q, Fu R Y,etal. 2009. Sequence analysis of 16S ribosomal DNA of phytoplasma associated with periwinkle yellows disease in Hainan. Acta Phytopathologica Sinica, 39(2): 212-216. [in Chinese]
Danet J L, Bonnet P, Jarausch W,etal. 2007.ImpandsecY, two new markers for MLST (multilocus sequence typing) in the 16SrX phytoplasma taxonomic group. Bulletin Insectol, 60(2): 339-340.
Dickinson M, Hodgetts J. 2013. Phytoplasma: methods and protocols. Totowa: Humana Press.
Firrao G, Martini M, Ermacora P,etal. 2013. Genome wide sequence analysis grants unbiased definition of species boundaries in “CandidatusPhytoplasma”. Syst Appl Microbiol, 36(8): 539-548.
Fox G E, Wisotzkey J D, Jurtshuk P. 1992. How close is close: 16S ribosomal RNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol, 42(1): 166-170.
Fraser C M, Read T D, Nelson K E. 2004. Microbial genome. Totowa: Humana Press.
Galdeano E, Torres L E, Menneguzzi N,etal. 2004. Molecular characterization of 16S ribosomal DNA and phylogenetic analysis of two X-disease group phytoplasmas affecting China-tree (MeliaazedarachL.) and Garlic (AlliumsativumL.) in Argentina. J Phytopathol, 152(3): 174-181.
Gundersen D E, Lee I M, Rehner S A,etal. 1994. Phylogeny of mycoplasmalike organisms (phytoplasmas): a basis for their classication. J Bacteriol, 176(17): 5244-5254.
Hodgetts J, Boonham N, Mumford R,etal. 2008. Phytoplasma phylogenetics based on analysis ofsecAand 23S rRNA gene sequences for improved resolution of candidate species of ‘CandidatusPhytoplasma’. Int J Syst Evol Microbiol, 58(8): 1826-1837.
Hogenhout S A, Oshima K, Ammar E D,etal. 2008. Phytoplasma: bacteria that manipulate plants and insects. Mol Plant Pathol, 9(4): 403-423.
Hu J X, Tian G Z, Lin C L,etal. 2013. Cloning expression and characterization of tRNA-isopentenyltransferase genes (tRNA-ipt) from paulownia witches’-broom phytoplasma. Acta Microbiologica Sinica, 53(8): 832-841. [in Chinese]
Jin K X, Tsai J H, Norris R C. 1982. Observation of bacterialike organisms and mycoplasmalike organisms inMeliaazedarachwith witches’-broom. Scientia Silvae Sinicae, 18(4): 422-424. [in Chinese]
Kube M, Schneider B, Kuhl H,etal. 2008. The linear chromosome of the plant-pathogenic mycoplasma ‘Candidatusphytoplasma mali’. BMC Genomics, 9: DOI:10.1186/1471-2164-9-306.
Lai F, Song C S, Ren Z G,etal. 2014. Molecular characterization of a new member of the 16SrV group of phytoplasma associated withBischofiapolycarpa(Levl.) Airy Shaw witches’-broom disease in China by a multiple gene-based analysis. Australian Plant Pathol, 43(5): 557-569.
Lee I M, Martini M, Bottner K D,etal. 2003. Ecological implications from a molecular analysis of phytoplasmas involved in a aster yellows epidemic in various crops in Texas. Phytopathol, 93(11): 1368-1377.
Lee I M, Gundersen D E, Davis R E,etal. 2004. ‘CandidatusPhytoplasma asteris’, a novel phytoplasma taxon associated with aster yellows and related diseases. Int J Syst Evol Microbiol, 54(4): 1037-1048.
Lee I M, Zhao Y, Bottner K D. 2006.SecYgene sequence analysis for finer differentiation of diverse strains in the aster yellows phytoplasma group. Mol Cell Probes, 20(2): 87-91.
Li Y, Piao C G, Tian G Z,etal. 2014. Multilocus sequences confirm the close genetic relationship of four phytoplasmas of peanut witches’-broom group 16SrII-A. J Basic Microbiol, 54(8): 818-827.
Li Y, Tian G Z, Piao C G,etal. 2005. Rapid molecular differentiation and identification of different phytoplasmas from several plants in China. Acta Phytopathologica Sinica, 35(4): 293-299. [in Chinese]
Maiden M C J, Bygraves J A, Feil E,etal. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA, 95(6): 3140-3145.
Maiden M C J. 2006. Multilocus sequence typing of bacteria. Annu Rev Microbiol, 60(60): 561-588.
Music M C, Pusic P, Fabre A,etal. 2011. Variability of stolbur phytoplasma strains infecting Croatian grapevine by multilocus sequence typing. Bulletin Insectol, 64(suppl): S39-S40.
Oshima K, Kakizawa S, Nishigawa H,etal. 2004. Reductive evolution suggested from the complete genome sequence of a plant-pathogenic phytoplasma. Nature Genet, 36(1): 27-29.
Saux M F. 2013. What new technologies bring to challenge of bacterial classification: unraveling evolution and ecology of plant pathogenic bacteria. Acta Phytopathologica Sinica, 43(suppl): 538.
Schneider B, Gibb K S, Seemüller E. 1997. Sequence and RFLP analysis of the elongation factor Tu gene used in differentiation and classification of phytoplasmas. Microbiol, 143(5): 3381-3389.
Shi B P, Li C L, Du Y J,etal. 2013. Molecular identification ofLactucasativayellows phytoplasma. J Shihezi University: Natural Science Edition, 31(6): 669-674. [in Chinese]
Tian G Z, Raychaudhuri S P. 1996. Paulownia witches’-broom disease in China: present status∥Raychaudhuri R S, Moromorosch K. Forest trees and palms: diseases and control. New Delhi: Oxford & IBH Publishing Company, 227-251.
Tran-Nguyen L T T, Kube M, Schneider B,etal. 2008. Comparative genome analysis of ‘Candidatusphytoplasma australiense’ (subgrouptuf-Australia I;rp-A) and ‘Ca. phytoplasma asteris’ strains OY-M and AY-WB. J Bacteriol, 190(11): 3979-3991.
Wei W, Davis R E, Lee I M,etal. 2007. Computer-simulated RFLP analysis of 16S rRNA genes: identification of ten new phytoplasma groups. Int J Syst Evol Microbiol, 57(8): 1855-1867.
Zhao Y, Wei W, Lee I M,etal. 2009. Construction of an interactive online phytoplasma classification tool,iPhyClassifier, and its application in analysis of the peach X-disease phytoplasma group (16SrIII). Int J Syst Evol Microbiol, 59(10): 2582-2593.
(责任编辑 王艳娜 郭广荣)
Multilocus Sequence Analysis for Revealing Finer Genetic Variation and Phylogenetic Interrelatedness of Phytoplasma Strains in 16SrI Group in China
Yu Shaoshuai Li Yong Ren Zhengguang Song Chuansheng Lin Caili Piao Chungen Tian Guozhong
(KeyLaboratoryofForestProtectionofStateForestryAdministrationResearchInstituteofForestEcology,EnvironmentandProtection,ChineseAcademyofForestryBeijing100091)
【Objective】The phytoplasmas in 16SrI group cause severe diseases of many crops and ecological plants in China. At present, the genetic variation and population structure of the yet-uncultured and very closely related phytoplasma strains in China are still not fully understood. A multilocus sequence analysis (MLSA) scheme was used to elucidate genetic diversity of phytoplasma strains in 16SrI group from different regions in China and their relationships with geographical distribution, and to compare the levels of variation in different housekeeping gene of different phytoplasma strains. The study aimed to provide some reference approach and evidence for finer detection, identification, classification and phylogenetic analysis of different phytoplasma strains in China. 【Method】 Ten housekeeping gene (rp,tuf,secA,secY,ipt,dnaK,fusA,gyrB,pyrGandrpoB) fragments combined with 16S rDNA sequences were employed to analyze the genetic variation and phylogenetic relationships of 18 phytoplasma strains infecting chinaberry, lettuce, mulberry, paulownia and periwinkle from ten provinces in China, using five phytoplasma strains, onion yellows phytoplasma (OY-M), aster yellows witches’-broom phytoplasma (AYWB),CandidatusPhytoplasma australiense (CPA), strawberry lethal yellows phytoplasma (SLY) andCandidatusPhytoplasma mali (CPM), of which whole genomes have been sequenced, as references. Sequence polymorphism and variation levels of different gene fragments were analyzed by multiple sequence alignment.【Result】 The nucleotide site polymorphisms ofrp,tuf,secA,secY,ipt,dnaK,fusA,gyrB,pyrGandrpoBgene fragments were used to resolve all strains into 15 sequence types (STs), demonstrating extensive genetic diversity among the 16SrI group strain population. All the strains, classified into 16SrI-B and -D subgroup by 16S rDNA analysis, were clustered into one clade and clearly differentiated into discrete subclades by phylogenetic analysis of the concatenated gene sequences. Ten chinaberry witches’-broom strains, which were most closely related to two mulberry dwarf strains and hardly distinguished with 16S rDNA, were definitely split into four distinct clusters and 8 STs apparently relative to their geographical origins. Two lettuce yellows strains in Sanming, Fujian province, China, were more closely related to the onion yellows OY-M strain in Japan than the periwinkle virescence and paulownia witches’-broom strains in China. The levels of variation indnaKlocus were higher than those in 16S rDNA and other genes tested.【Conclusion】 It is suggested that the MLSA should potentially be a useful and reliable approach for phytoplasma identification and differentiation as well as for depth examination of strain diversity and this method can be wildly used to study the genetic variation and evolutionary relationships of phytoplasma strains among various groups or subgroups in the future.
phytoplasma; multilocus sequence analysis (MLSA); genetic variation;sequence type (ST); phylogenetic evolution
10.11707/j.1001-7488.20170312
S763.11
A
1001-7488(2017)03-0105-14
Received date: 2016-02-02; Revised date: 2016-03-10.
Funded project: The National High Technology Research and Development Program of China (2012AA101501); National Infrastructure of Microbial Resources of China (NIMR2014-7).
* Corresponding author: Tian Guozhong.
Acknowledgements: The authors thank Prof. Fan Zaifeng in China Agricultural University for his kind assistance in writing, Prof. Kuai Yuanzhang in Jiangsu University of Science and Technology, Liu Jianmi, Yan Shuping and Lin Jixiu in Plant Protection and Quarantine Station of Sanming City for providing the phytoplasma strains.
