黔产大菟丝子中绿原酸的定性与定量分析
2017-04-24冯华石尚友罗秀琼王世俊郑凰雅王
冯华+石尚友+罗秀琼+王世俊+郑凰雅+王祥培
【摘要】目的:对黔产大菟丝子中绿原酸进行定性与定量分析。方法:采用TLC色谱法,以绿原酸为对照,建立定性鉴别方法;采用HPLC色谱法,建立其含量测定方法。结果:TLC法能很好分离各成分;高效液相色谱法绿原酸在00102~01142mg范围内与峰面积响应值呈良好的线性关系(r=09998),平均加样回收率为 990%。结论:本法简便可行,重复性好,为评价大菟丝子质量提供实验依据,可用于大菟丝子药材的有效鉴别。
【关键词】黔产大菟丝子;绿原酸;定性与定量
【中图分类号】R284【文献标志码】 A【文章编号】1007-8517(2017)05-0014-03
大菟丝子为旋花科植物大菟丝子(Cuscuta Japonica Choisy)的干燥成熟种子,收载于2003版贵州省《中药材、民族药材质量标准》[2]。本品为少数民族药材,性辛、甘、平,具有滋补肝肾、固精缩尿、安胎、明目、止泻功效,临床用于肾虚腰痛、阳痿遗精、目昏耳鸣、胎动不安、尿频等。大菟丝子药材中含新绿原酸、绿原酸、隐绿原酸、咖啡酸、3,4-二咖啡酰奎宁酸、3,5-二咖啡酰奎宁酸、4,5-二咖啡酰奎宁酸等有机酸类成分[3],其中绿原酸具有抗病毒、保肝利胆作用。笔者查阅相关文献资料,目前尚无对大菟丝子中绿原酸薄层鉴别和HPLC法含量测定方法的报道,笔者对大菟丝子中绿原酸薄层和含量测定进行研究,建立了绿原酸的薄层鉴别与含量测定方法,为大菟丝子药材质量标准提升和控制提供参考。
1仪器与材料
11仪器Agilent LC-1260高效液相色谱议(包括四元泵,DAD,柱温箱,自动进样器,工作站);AB204-S型梅特勒电子天平;KQ-500DV型数控超声波清洗机(昆山市超声仪器有限公司)。
12材料绿原酸对照品(批号:110753-20314,中国食品药品检定研究院)。聚酰胺薄膜(购于青岛海洋化工有限公司),乙睛为色谱纯,水为纯净水,其它试剂均为分析纯。
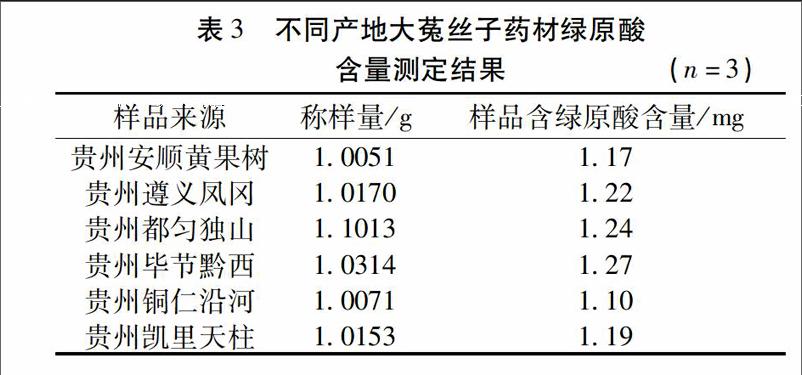
大菟丝子采集于贵州省各地区,经遵义市食品药品检验所邓顺超副主任中药师鉴定为旋花科植物大菟丝子的干燥成熟种子,样品来源见表1。
2 方法与结果
21薄层鉴别[1]取本品粉末约10g,置具塞量瓶中,加甲醇10mL,超声30min,滤过,滤液水浴上浓缩约1mL,作为供试品溶液。另取绿原酸对照品,加甲醇溶液制成每1mL含01mg的溶液,作为对照品溶液。照薄层色谱法(中国药典2015年版四部附录)试验,吸取上述两种溶液各5μL,分别点于同一聚酰胺薄膜板上,以甲醇-冰醋酸-水(4∶1∶5)为展开剂,展开,取出,晾干,喷以三氯化铝乙醇溶液,置紫外光燈(365nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。结果表明,分离度好,斑点显色清晰,阴性无干扰,见图1。
22绿原酸的含量测定[3]
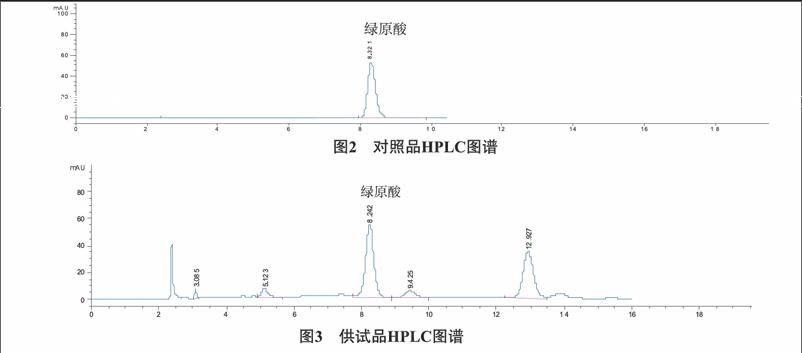
221色谱条件[2-3]色谱柱:Eclipse XDB-C18(5μm,46mm×250mm);流动相:乙睛-01%磷酸溶液(12∶88),流速为:10mL/min,柱温30℃,检测波长为325nm。进样量10μL,分离度为24,理论塔板数按绿原酸峰计,应不低于3500,色谱图见图2、图3。
222对照品溶液的制备精密称取绿原酸对照品1060mg,置25mL容量瓶中,加甲醇溶解至刻度,摇匀,精密吸取2mL置10mL容量瓶中,加甲醇至刻度,摇匀,即得。
223供试品溶液制备精密称取大菟丝子药材粉末(过二号筛)约10g,置具塞瓶中,加甲醇25mL,称定重量,超声50min,放置至室温,用甲醇补足重量,过滤,取续滤液,即得。
224检测波长确定照紫外-可见分光光度法,在200~400nm的波长范围内对对照品溶液、供试品溶液以及阴性对照品溶液进行扫描。结果显示,绿原酸和样品在325nm波长处有最大吸收,因此选择检测波长为325nm。
225线性关系的考察分别配制6个不同浓度绿原酸对照品溶液,浓度分别为00102、00204、00408、00816、00979、01142mg/mL,进样量为10μL,注入高效液相色谱仪,测定峰面积,以绿原酸浓度(mg)为横坐标,峰面积积分值(A)为纵坐标,计算绿原酸回归方程为:Y=68654X+00560(r=09998)。结果表明,在00102~01142mg范围内线性关系良好。
226精密度试验精密吸对照品溶液10μL,注入高效液相色谱仪中,测定6份,平均峰面积657836,RSD=04%,表明仪器精密度良好。
227稳定性试验取同一批药材粉末约10g,精密称定,按供试品溶液项下方法制备供试品溶液,精密吸取供试品溶液10μL,注入高效液相色谱仪中,分别在0、2、4、8、12h测定一次,平均峰面积为682360,RSD为08%,表面供试品溶液在12h内稳定。
228重复性试验取同一批药材粉末约10g6份,精密称定,照“223”项下制备方法制备成供试品溶液。进样10μL,平均含量为012%,RSD为11%,表明重复性良好。
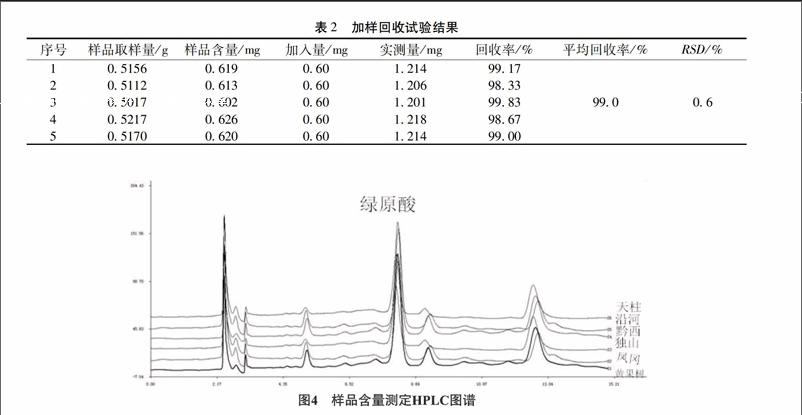
229加样回收试验称取已测定S1号样品粉末5份(含量为012%),每份约05g,精密称定,加入绿原酸对照品浓度为030mg/mL溶液2mL,按“223”项下的方法制备供试品溶液,在“221”项下色谱条件进样测定,计算绿原酸平均回收率为990%,RSD为06%,结果见表2。
2210含量测定分别测定大菟丝子药材不同产地样品,测定其含量,计算结果见表3,图谱见图4。
3讨论
31绿原酸的TLC鉴别采用乙醇、甲醇溶剂提取后,乙醇提取溶液的斑点分离效果不佳,甲醇提取的各斑点能有效分离。按正文方法操作,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。置紫外光灯(365nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。结果表明,分离度好,斑点显色清晰。以甲醇-冰醋酸-水不同比例为展开剂,在聚酰胺薄膜上展开,经过不同比例调整。以甲醇—冰醋酸-水(4∶1∶5)溶液为展开剂,在聚酰胺薄膜上展开,斑点清晰,分离完全,。所以选择聚酰胺薄膜板,甲醇—冰醋酸-水(4∶1∶5)溶液为展开剂。
32绿原酸提取条件的优选选用甲醇、80%甲醇、50%甲醇作为提取溶剂,超声、回流提取时间为40、50、60min。回流与超声提取绿原酸含量没有差异,选择甲醇25mL超声50min,药材中绿原酸提取完全,选用甲醇为溶剂比较好;对色谱条件进行选择,以乙睛-01%磷酸溶液(12∶88),流速为10mL/min;柱温30℃;检测波长为325nm,各组分峰达到完全分离。
综上,本法简便、快速,准确度高,可用于大菟丝子的质量控制。
参考文献
[1]国家药典委员会.中华人民共和国药典(四部)[M].北京:中国医药科技出版社,2015:57.
[2]贵州省食品药品监督管理局编.《贵州省中药材、民族药材质量标准》[S].贵阳:贵州科技出版社,2003:39.
[3]瞿燕,李琼, 魏文龙,等.大菟丝子生品和盐炙品HPLC-UV特征图谱及7个有机酸含量变化研究[J].药物分析,2014,34(5):824-829.
