福岛核事故释放放射性气溶胶的化学分析进展
2017-04-21龙兴贵
谢 波,胡 胜,龙兴贵
中国工程物理研究院 核物理与化学研究所,四川 绵阳 621999
福岛核事故释放放射性气溶胶的化学分析进展
谢 波,胡 胜,龙兴贵
中国工程物理研究院 核物理与化学研究所,四川 绵阳 621999
本文综述了针对日本福岛核电站(FDNPP)事故后,早期阶段释放的放射性气溶胶的化学分析工作进展。内容主要涉及负载铯、钚、氙、碘等核素的气溶胶粒子。从粒子的取样与样品处理、粒子特性分析与表征、粒子输运与沉降行为、同类事故比较等角度,总结了事故后放射性气溶胶的部分物化特性与动力学行为。
气溶胶;放射性核素;核事故;化学分析
2011年3月11日,日本福岛核电站(FDNPP)发生重大事故,释放的大量放射性物质穿越了整个北半球,事故等级达到国际核事件分级表的最高级——7级[1],直接造成全球核能形势的深刻变化,国际社会和公众对核能安全的关注度和敏感度显著增加,对核安全标准的审核和修订、核设施的安全性和可靠性、核安全监管的有效性和透明度提出了更为严格的要求。在这5年间,诸多科学机构对福岛核事故产生的放射性气溶胶开展了大量研究,主要涉及到模型计算、物理测量与化学分析三个方面。本文综述了针对事故早期阶段释放的放射性气溶胶而开展的化学分析与表征工作,重点涉及到铯、钚、氙、碘等多种放射性核素。各国研究的目的在于描述真实气溶胶的化学与物理性质(包括化学形式与核素组成、粒子尺寸分布与形状、相态与晶体结构、水溶性、滞留时间、电荷特征、动力学演变等信息),预防次生事故发生,寻找有效方式去除环境中的放射性物质和阻止气溶胶的再悬浮,准确评估各种数值模型的改进和事故后人体照射风险,揭示事故厂区的具体真相。同时,通过对福岛核事故后放射性气溶胶化学分析现状的调研,期望对放射性气溶胶的相关研究有所帮助。
1 铯-负载粒子气溶胶
1.1 粒子的取样与处理
在日本筑波气象学研究所,来自不同研究机构的Adachi[2]和Abe[3]等分别使用大容量气溶胶取样器(1 000 m3/d)及石英纤维材料收集了包含放射性材料的气溶胶,一个成像板(imaging plate, IP)和微操纵器(micro-manipilator)用于过滤材料上探测和分离放射性粒子。在同一批次的样品中,每个样品大约有100个小点(每一个小点都表示放射性材料的存在),如图1[2]所示,换算成近似粒子浓度大约为每立方米10个放射性粒子。
因为使用扫描电镜(SEM)探测放射性粒子,必须减少过滤器上非放射性粒子的数目。Adachi等[2]的处理是将取样滤纸裁剪成许多小块,使用IP和锗探测器测量每一个小块,结果发现了3个放射性的Cs-负载粒子。然后描述粒子的形状、组成、水溶性和尺寸,评价了它们的形成过程、环境和潜在的健康影响。而Abe等[3]从过滤材料上取了3种粒子放在玻璃基片上,分别命名为A、B、C,开展比对测量。
1.2 分析与表征
样品的分析与表征采用了多种手段。首先是扫描电镜-能量分散谱(SEM-EDS)分析,对样品进行可视显微观察的同时,进行材料表面元素的成分分析和元素自动识别;然后是γ射线能谱测量和针对重元素的X射线(XRF)荧光分析,并以高灵敏度特征的激光烧蚀-电感耦合等离子体质谱(LA-ICP-MS)为辅助测量手段;最后进行同步辐射-X射线微束分析(包括SR-μ-XANES和SR-μ-XRD),前者用于评价粒子的形成条件和过渡元素的化学状态分析,后者用于揭示微粒的晶体结构。这些分析技术的综合运用,目的是揭示来自于FDNPP事故的Cs承载微粒的本质特征。
Adachi等[2]发现样品的放射性浓度在2011年3月14—15日和3月20—22日有两个峰值,与Kinoshita[4]的观点一致,认为气象学条件(例如雨和风向)和放射性粒子的释放是高表面沉积事件的主要原因。滤纸上放射性物质的分布是多斑点的、参差不齐的,放射性粒子的数目相对少,但活度水平相对较强。单个Cs-负载粒子的分析示于图2。由图2可知,每一块碳胶上的Cs-负载粒子都是球形,直径可以测量,EDS谱图显示有Cs峰,Cs分布的元素成像显示,球形粒子由Cs伴随大量的Fe、Zn和少量的Cl、Mn、O组成。Cs粒子的衰变校正活度对137Cs和134Cs是不同的,若假设了粒子密度,则可以从Cs的活度推算出它在粒子中的质量百分比,以及大于某种尺寸粒子的平均粒子数量浓度[5]。此外,Cs粒子水溶性的分析表明,粒子接触水后形状未发生变化。

图1 2011年不同时间段过滤器样品上放射性材料分布的IP结果[2]Fig.1 Distribution of radioactive materials on the filter samples measured with the IP at different time in 2011[2]
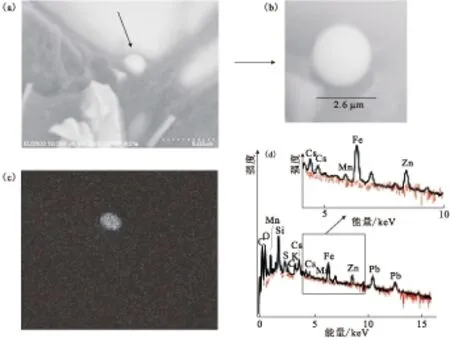
(a)——一个Cs-负载粒子被埋置在碳胶中,(b)——球形的Cs-负载粒子,(c)——Cs-负载粒子的元素成像,(d)——粒子的能量分散谱图图2 单个Cs-负载粒子的分析[2]Fig.2 Analysis of single Cs-bearing particle[2]
Abe等[3]认为,同步辐射-X射线微束分析可以揭示福岛事故期间释放的放射性微粒的化学性质细节,可以更好地明白事故早期厂区究竟发生了什么。他在收集的气溶胶样品中发现了包含放射性Cs的三种球形微粒,利用SR-μ-X射线荧光分析探测到11种元素:Fe、Zn、Rb、Zr、Mo、Sn、Sb、Te、Cs、Ba和Pb,并与SEM结果高度一致。粒子中除了Pb元素,其它元素都是均相分布,尽管谱图中有Pb-L线的强峰,但Pb的分布与其它元素和SEM结果有明显区别,表明Pb不是来源于粒子成分。此外,其中两种粒子中还第一次发现了U,并被U的L-边缘X射线吸收近边结构(L-XANES)光谱所证实,表明U燃料和它的裂变产物连同放射性Cs一起被保留在这些粒子中。Abe等[3]的研究结果说明,FDNNP受损程度足以说明安全壳以气溶胶粒子的形式释放U燃料和裂变产物。更为重要的是,Fe、Zn、Mo、Sn的SR-μ-XANES分析显示,峰的位置和形状与玻璃参照材料一致,元素以Fe3+、Zn2+、Mo6+和Sn4+的形式出现。而在SR-μ-XRD中采用硅粉作为参照材料时,硅粉显示了清晰的Debye-Scherrer环,但粒子没有衍射峰。这些晶体结构的研究结果说明,粒子是非晶形的、玻璃态材料,加上自身的小球形状,说明微粒在氧化条件下经历了高温熔融、迅速冷却的过程后形成气溶胶。
众所周知,作为铀核裂变反应的结果,裂变产物有9种元素(Rb、Ag、Zr、Mo、Sn、Sb、Te、Cs、Ba)可以在微粒中找到[6]。由于熔融堆芯可能与混凝土反应,粒子中可能存在Si。由于缺乏对反应堆受损状态的评估,无直接证据表明这些元素的起源,只能推断铀燃料、裂变产物和反应堆构件可能是元素的起源,它们在事故期间熔融在一起而形成球形微粒。
购买干货、零食时,最好到规模大、信誉好、食品质量把关较严的商场或超市选购;包装食品要注意查看品名、厂家、保质期等,特别要观察是否新鲜,是否在保质期内,包装是否完整无损、有无鼓包(涨袋)及霉变等现象。不要购买和食用“三无”食品。
1.3 输运与沉降行为
尽管福岛核事故造成了全球影响,但在相当长的时间内仍然不知道反应堆究竟发生了什么,释放的放射性Cs的值也从9 PBq至36 PBq不等,不知道释放到环境中放射性物质的化学与物理性质、粒子的输运与沉降行为[7],日本核工业安全委员会2011年6月递交给国际原子能机构(IAEA)的报告[8]称,137Cs的释放可能小于20 kg,福岛事故造成的137Cs尘降量只有切尔诺贝利的17%,总剂量只有切尔诺贝利的5%~6%。放射性物质的全球性扩散,很难依靠纯化学方式进行探测[9]。有模拟结果显示[10],Cs-负载粒子落到地面主要是干式沉降,少量放射性Cs附着于其它占统治地位的粒子上,例如硫酸盐粒子作为137Cs的载带介质,SEM-EDS分析也证实了普遍存在于气溶胶样品中的大量硫酸盐和矿物粉尘。Kaneyasu等[11]测量了事故后47 d的134Cs和137Cs气溶胶活性尺寸分布,发现第一批样品(2011年4月28日至5月12日)的空气动力学直径中间值(AMAD)分别为0.54 μm和0.53 μm,但是在第二批样品(2011年5月12日至26日)中均为0.63 μm。这些放射性Cs的活性尺寸分布属于积聚模态,与非海盐的硫酸盐气溶胶的质量尺寸分布几乎重叠。硫酸盐是潜在的放射性核素的输运介质,而再悬浮土壤颗粒不是主要的空中放射性物质。这也就解释了为什么在切尔诺贝利事故后不同监测点获得了相似的放射性Cs的活性尺寸,结果示于图3[11]。
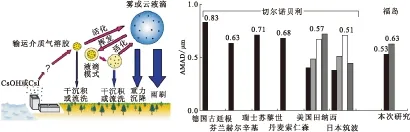
图3 硫酸盐气溶胶作为放射性Cs的输运介质[11]Fig.3 Sulfate aerosol as a transport medium of radiocesium[11]
Stohl等[12]对137Cs使用了大气活度浓度测量和大体积沉积测量,反演结果给出了总释放量为36.6 PBq(不确定度范围为20.1~53.1),约为切尔诺贝利事故释放量的43%,远远高于日本核工业安全委员会递交的报告。当向福岛核电站4号机组乏燃料池喷洒水时,数值出现数量级式的突然下降,这表明释放不仅仅是来源于受损的堆芯,也来源于4号机组的乏燃料池。估计6.4 PBq的137Cs(占2011年4月20日前的总沉降物的18%)沉积在日本陆地表面,剩余部分大多落在北太平洋,仅仅0.7 PBq沉降在非日本的陆地表面。
1.4 微粒对环境的影响
粒子中一些重元素的产生是由于核裂变反应,如果这个假设是正确的话,当它们在事故期间释放时,很可能包含额外的短寿命放射性核素。因此,这些粒子的比活度比Cs高数倍,过去的研究[13]报道这些粒子是难溶于水的,揭示它们是高氧化价态下的玻璃态材料,球形Cs-负载粒子在陆地表面的滞留时间比水溶性Cs粒子长[14]。这些特征表明,它们对环境的影响是长期的。
日本东京技术研究所和东京海洋与技术大学[15]提供了福岛事故周边放射性核素分布的详细地图和放射性核素在环境中的沉积与迁移。他们认为:(1) 陆地表面的放射性核素是释放到大气的挥发性裂变产物的沉积所造成,主要是137Cs和131I;气溶胶粒子的反弹与再悬浮进入大气,造成热点地区污染程度和位置的改变;(2)131I与134Cs和137Cs原子比率的变化表明,事故气溶胶的沉积造成了海水放射性的变化,推断气溶胶可以到达1 900 km之外的北太平洋监测站点。Burns等[16]认为,在堆芯熔融事故中,挥发性裂变产物的气态释放是瞬间发生的,大气扩散有助于缓解这些释放对环境的影响。
2 钚气溶胶
2.1 环境样品的收集与放化流程
大多数环境样品的收集方式是,收集表面土壤样品和枯枝落叶层(litter layer)样品用于钚同位素的定量分析,主要分析仪器是扇形场ICP-MS。有数据表明[17],到2011年5月样品收集结束时,释放的钚沉积在枯枝落叶层,尚未达到它下面的表面土壤。
2.2 钚气溶胶分析结果
基于ORIGEN模型的模拟[18]和环境样品分析数据,Zheng等[19]全面总结和分析了福岛事故中钚同位素的释放,结果示于图4。分析主要集中于4个方面:(1) 钚同位素的环境分布;(2) 源项钚同位素组成;(3) 受损反应堆单元或乏燃料池钚释放的源项;(4) 钚释放总量。结果表明,痕量的钚同位素(约占堆芯存量的2×10-5%)释放到环境中,全部来源于受损的反应堆,而不是乏燃料池。至于海洋污染,沿福岛的西太平洋海岸30 km范围内未发现额外的钚。Lujaniene等[20-21]用质谱分析了在立陶宛收集到的钚气溶胶样品(从2011年3月23日至4月15日),得到了240Pu/239Pu原子比率和238Pu/239+240Pu活度比率。Shinonaga等[22]利用加速器质谱(AMS)和ICP-MS分析了距离福岛120 km事故前、后的大气钚气溶胶样品和非天然铀气溶胶样品,测量了240Pu/239Pu、241Pu/239Pu、234U/238U、235U/238U、236U/238U等多个原子比率。在相同测量条件下,AMS探测器计数率粗略地与离子源样品中的钚浓度成比例,5个样品的239Pu计数率大于0.07 s-1,其它样品在0.02~0.04 s-1,假设样品是完全等分的,239Pu活度浓度的计算值约为130 nBq/m3。这些结果说明,钚和非天然铀在事故后的数天内被气溶胶和风输运了120 km,与核事故造成的放射性核素总剂量相比,钚的贡献可以忽略不计。

图4 福岛核事故钚气溶胶的释放[19]Fig.4 Discharges of Pu aerosol from Fukushima Daiichi nuclear accident[19]
2.3 与其它钚释放事件的比较
与切尔诺贝利核电站事故相比,福岛核事故的钚有更高的241Pu/239Pu原子比率,但是240Pu/239Pu的比率更低一些。然而,由于切尔诺贝利事故释放了大约8.7×1013Bq的239+240Pu,241Pu/239+240Pu的活度比率要比福岛核事故的低很多。钚原子比率随反应堆燃料燃耗时间的增加而增加,相对高的241Pu/239+240Pu活度比表明3号反应堆受到了损害,因为3号堆是混合堆芯,包含铀燃料与混合的铀-钚氧化物燃料(钚氧化物占6%)。为了理解切尔诺贝利事故钚释放与福岛事故的区别,有研究[17]估算了大气释放钚量和堆芯库存释放的比例,比较了长崎原子弹爆炸、全球尘降与福岛核事故的钚同位素活度比率,发现距离福岛核电站数公里和数十公里的地方,241Pu/239Pu的比率比大气核武器测试期间的数值还要高很多。
3 吸附133Xe的气溶胶
与气溶胶绑定的137Cs相比,惰性气体133Xe具有完全不同的释放特征,福岛事故中总释放量达到15.3 EBq,超过切尔诺贝利事故两倍,是历史上最大规模的放射性惰性气体释放[12]。为了测定放射性核素释放与高度和时间的关系,研究方式一般是基于燃料库存和文献事故记载,先进行第一次释放速率的猜测,然后猜测被反演模拟改进,改进的方式是考虑大气输运模型FLEXPART和分布在日本、北美等地区的几十个监测站的测量数据。事实上,各国估计的133Xe释放量总和高于福岛核电站133Xe的库存总量,可能的原因是133I(半衰期为20.8 h)的衰变形成了133Xe。
有证据显示[23],在第一次排气之前133Xe就开始释放,表明反应堆组件由于超压出现结构性的破坏和泄漏。对包括日本和整个北半球在内的放射性云的扩散和沉积模式的探测研究表明[24],受西风影响,2011年3月15日放射性烟云到达北美,3月22日到达欧洲,到4月中旬,133Xe均匀分布在整个北半球的中部纬度,而且第一次被南半球的澳大利亚达尔文站监测到。
太平洋西北实验室(PNNL)采用瑞典SAUNA(Swedish Automatic Unit for Noble Gas Ac-quisition)系统[25],在3月16日(事故后第4天)第一个监测到短寿命裂变产物133Xe浓度的变化,监测点距离福岛7 000 km,133Xe浓度最大值超过40 Bq/m3,超过正常平均值的4万倍[26]。氙的收集与测量示于图5[25],包含的流程有:(1) 用气相色谱仪测量样品中氙总量,确定空气取样量;(2) 用β-γ符合系统测量氙同位素活度和氙总量,测量完成后样品再返回样品归档瓶中;(3) 用转移装置将样品从归档瓶转移至炭纤维源盒,用气相色谱确定其转移效率;(4) 用锗γ谱仪测量氙同位素活度,最后得到气溶胶吸附的氙同位素活度浓度。释放放射性核素的大气输运依赖于源项高度,可以将释放分为3层,分别距离地面的高度范围是0~50 m、50~300 m、300~1 000 m。这种方式与朝鲜2006年10月9日宣称核试验后,Ringbom等[27]在韩国境内测量空气中放射性氙的地面水平的方式一致。

图5 瑞典SAUNA系统用于133Xe的收集与测量[25]Fig.5 SAUNA system for sampling and analysis of 133Xe [25]
4 其他气溶胶核素
从核事故中释放的放射性粒子种类繁多(福岛事故已经监测到放射性核素73种[28]),其放射学的重要性不仅依赖于放射性核素和它的化学形式,也依赖于粒子的空气动力学性质(例如尺寸、形状和密度)。放射性气溶胶的粒子尺寸分布和粒子与其它气溶胶组分相混合的状态,对于理解它们的形成、输运和沉积来说,是非常重要的参数。
Thakur等[29]总结了福岛事故后北半球放射性核素测量的进展,收集到碘核素粒子的几何尺寸直径范围是0.01~10 μm,超过80%的131I为精细气溶胶粒子(0.01~1 μm),不足20%的是粗粒子(>10 μm)。131I活度最大时的粒子直径是在0.1~0.36 μm,活度最小的粒径范围是0.36~1.8 μm。此结果与Paatero等[30]对切尔诺贝利事故释放的、携带131I气溶胶粒子直径的测量结论一致,与137Cs、132Te、103Ru相比(0.63~0.93 μm),131I倾向于附着更小的气溶胶微粒(0.33~0.57 μm)。而捷克国家辐射防护研究所的Malá等[31]详细比对了福岛事故与切尔诺贝利事故中放射性气溶胶的粒子尺寸分布,同样认为,切尔诺贝利事故释放的放射性核素根据AMAD分为两种类型:难溶放射性核素(140Ba、140La、141Ce、144Ce、95Zr和95Nb)与挥发性核素(134Cs、137Cs、103Ru、131I和132Te),前者的AMAD大约是后者的3倍以上,并且挥发性核素AMAD的平均值(0.51 μm)与福岛事故的十分接近。以此为数据基础,文献[32-33]给出了欧洲上空放射性核素活度的时空分析结果,基本上反映了福岛核电站的受损状况。
Burns等[16]在比较了3次商业核电站事故后认为,1979年的美国三哩岛事故,由于原始安全壳泄漏,放射性裂变产物气体大量释放,但没有分散的燃料粒子;1986年的切尔诺贝利事故,放射性材料的爆炸性释放,裂变气体和挥发性裂变产物(I、Cs)被大量释放,大约6 t燃料变为大气介质粒子进行传播;2011年福岛事故尽管大部分燃料是UO2,但有32个混合氧化物燃料组件包含钚,相对于大量放射性的海水,气体和挥发性裂变产物的放射性是少量的。Burns等[16]还总结了不同类型的放射性材料从事故核燃料中得到释放,其中提到了氙、氪在燃料颗粒内形成了分散性好的气泡,金属裂变产物(Mo、Tc、Ru、Rh、Pd)作为微米或纳米尺寸的非混相粒子分散在燃料中,挥发性元素(I、Cs)迁移进入燃料颗粒的边缘与裂缝,燃料靶丸非均匀地燃耗导致靶丸边缘有更高的239Pu浓度。有研究[34]显示,辐照燃料温度在2 530 ℃时,释放部分中90%是Cs、I和惰性气体等裂变产物,5%是Mo,小于1%是Sr,锕系的释放非常低。
Kinoshita等[35]对事故后日本中东部地区放射性核素的分布进行了评估。通过氢爆炸、排气和泄漏方式释放传播的裂变产物种类繁多,除了134Cs、136Cs、137Cs和131I,高挥发性核素还包括129Tem,这些放射性核素被空气包(air parcel)载带,随后通过湿式和干式沉淀累积在地面上,大多数气溶胶和水溶性气体被雨水冲刷。依据HYSPLIT(Hybrid Single Particle Lagrangian Integrated Trajectory)模型[36]、气象数据[37]、全球数据同化系统(GDAS)和地理条件[38],分析出空气包的走向,确定这些核素的具体沉积位置,而非挥发性元素则被气溶胶吸附,粒子和气体状态的识别只能依靠大气中的碘[39]。HYSPLIT模型只是福岛核事故后催生的众多计算模型[40]中的一种,各类模型的研究进展将在另外的论文中描述。
5 结论与展望
福岛核事故后放射性气溶胶的化学分析结果表明:(1) 福岛核事故释放的放射性核素污染,跨越了整个北半球;(2) 负载铯、碘等挥发性核素气溶胶、铀钚气溶胶和吸附氙的气溶胶是核事故后重点关注的对象,它们的物化特性与动力学行为存在很大差异;(3) 放射性气溶胶的释放特征完全不同于切尔诺贝利核事故。
从上述放射性气溶胶化学分析的现状可以看出:(1) 代表性的气溶胶取样手段、严谨的同位素放化分离流程、先进的仪器分析技术、紧密关联的物理测量辅助和数值模型,是放射性气溶胶化学分析成败的关键,直接制约着气溶胶源项特性研究水平的高低,从而影响着核事故过程、人员风险和环境威胁评估的可靠性;(2) 福岛核事故产生的大量放射性气溶胶提供了非常宝贵的研究机遇,有力推动了气溶胶科学技术的发展;(3) 对放射性核素的释放与输运、照射途径还缺乏足够的、定量的理解,大多数分析研究还集中于研究未损坏燃料的放射性核素释放,推断极端条件下,例如发生堆芯熔融、氢气爆炸、强辐射场等情形下的气溶胶释放特征与动力学行为将是一个令人瞩目的研究方向。
[1] International Nuclear Event Scale. The international nuclear and radiological event scale user’s manual[M]. Vienna: International Atomic Energy Agency, 2008.
[2] Adachi K, Kajino M, Zaizen Y. Emission of spherical cesium-bearing particles from an early stage of the Fukushima nuclear accident[J]. Sci Rep, 2013, 3: 2554.
[3] Abe Y, Iizawa Y, Terada Y. Detection of uranium and chemical state analysis of individual radioactive microparticles emitted from the Fukushima nuclear accident using multiple synchrotron radiation X-ray analyses[J]. Ana Chem, 2014, 86: 8521-8525.
[4] Kinoshita N. Assessment of individual radionuclide distributions from the Fukushima nuclear accident covering central-east Japan[J]. Proc Nat Acad Sci USA, 2011, 108: 19526-19529.
[5] Yasunaria T J, Stohlb A, Hayano R S. Cesium-137 deposition and contamination of Japanese soils due to the Fukushima nuclear accident[J]. Proc Nat Acad Sci USA, 2011, 108, 49: 19530-19534.
[6] Saunier O, Mathieu A, Didier D, et al. An inverse modeling method to assess the source term of the Fukushima Nuclear Power Plant accident using gamma dose rate observations[J]. Atoms Chem Phys, 2013, 13(6): 15567-15614.
[7] Golovnin I S. Properties of plutonium dioxide as nuclear fuel[J]. Atom Ener, 2000, 89(2): 627-637.
[8] Cyranoski D, Brumfiel G. Fukushima impact is still hazy: chaos and bureaucracy hamper assessment of nuclear crisis[J]. Nature, 2011, 477: 139-140.
[9] Morino Y, Ohara T, Watanabe M, et al. Episode analysis of deposition of radiocesium from the Fukushima Daiichi Nuclear Power Plant accident[J]. Environ Sci Technol, 2013, 47: 2314-2322.
[10]Koizumi A, Harada K H, Niisoe T, et al. Preliminary assessment of ecological exposure of adult residents in Fukushima prefecture to radioactive cesium through ingestion and inhalation[J]. Environ Health Prev Med, 2012, 17: 292-298.
[11]Kaneyasu N, Ohashi H, Suzuki F, et al. Sulfate aerosol as a potential transport medium of radiocesium from the Fukushima nuclear accident[J]. Environ Sci Techol, 2012, 46: 5720-5726.
[12]Stohl A, Seibert P, Wotawa G, et al. Xenon-133 and caesium-137 releases into the atmosphere from the Fukushima Daiichi Nuclear Power Plant: determination of the source term, atmospheric dispersion, and deposition[J]. Atmos Chem Phys, 2012, 12: 2313-2343.
[13]Kanai Y. Monitoring of aerosols in Tsukuba after Fukushima Nuclear Power Plant incident in 2011[J]. J Environ Radioact, 2011, 111: 33-37.
[14]Tagami K, Uchida S, Uchihori Y, et al. Specific activity and activity ratios of radionuclides in soil collected about 20 km from the Fukushima Daiichi Nuclear Power Plant: radionuclide release to the south and southwest[J]. Sci Total Environ, 2011, 409: 4885-4888.
[15]Yoshida N, Kanda J. Tracking the Fukushima radionuclides[J]. Science, 2012, 336: 1115-1116.
[16]Burns P C, Ewing R C, Navrotsky A. Nuclear fuel in a reactor accident[J]. Science, 2012, 335: 1184-1187.
[17]Zheng J, Tagami K, Watanabe Y, et al. Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident[J]. Sci Rep, 2012, 2: 304.
[18]Schwantes J M, Orton C R, Clark R A. Analysis of a nuclear accident: fission and activation product releases from the Fukushima Daiichi nuclear facility as remote indicators of source identification, extent of release, and state of damaged spent nuclear pool[J]. Environ Sci Technol, 2012, 46: 8621-8627.
[19]Zheng J, Tagami K, Uchida S. Release of plutonium isotopes into the environment from the Fukushima Daiichi Nuclear Power Plant accident: what is known and what needs to be known[J]. Environ Sci Technol, 2013, 47: 9584-9595.
[20]Lujianiene G, Bycenkiene S, Povinec P P, et al. Radionuclides from the Fukushima accident in the air over Lithuania: measurement and modeling approaches[J]. J Environ Radioact, 2012, 114: 71-80.
[21]Lujianiene G, Valiulis D, Bycenkiene S, et al. Plutonium isotopes and241Am in the atmosphere of Lithuania: a comparison of different source terms[J]. Atmos Environ, 2012, 61: 419-427.
[22]Shinonaga T, Steier P, Lagos M, et al. Airborne plutonium and non-natural uranium from the Fukushima DNPP found at 120 km distance a few days after reactor hydrogen explosions[J]. Environ Sci Technol, 2014, 48: 3808-3814.
[23]Champion D, Korsakissok I, Didier D, et al. The IRSN’s earliest assessments of the Fukushima accident’s consequences for the terrestrial environment in Japan[J]. Radioprotection, 2013, 48(1): 11-37.
[24]Wetherbee G A, Gay D A, Debey T M, et al. Wet deposition of fission-product isotopes to North America from the Fukushima Daiichi incident, March 2011[J]. Environ Sci Technol, 2012, 46: 2574-2582.
[25]Ringbom A, Larson T, Axelsson A, et al. SAUNA—a system for automatic sampling, processing, and analysis of radioactive xenon[J]. Nuclear Instruments & Methods in Physics Research Section A, 2003, 508(3): 542-553.
[26]Bowyer T W, Biegalski S R, Cooper M, et al. Elevated radioxenon detected remotely following the Fukushima nuclear accident[J]. J Environ Radioact, 2011, 102: 681-687.
[27]Ringbom A, Elmgren K, Lindh K, et al. Measurements of radioxenon in ground level air in South Korea following the claimed nuclear test in North Korea on October 9[J]. J Radioanal Nucl Chem, 2006, 282(3): 773-779.
[28]Mathieu A, Korsakissok I, Quélo D, et al. Atmospheric dispersion and deposition of radionuclides from the Fukushima Daiichi Nuclear Power Plant accident[J]. Elements, 2012, 8: 195-200.
[29]Thakur P, Ballard S, Nelson R. An overview of Fukushima radionuclides measured in the northern hemisphere[J]. Sci Total Environ, 2013, 458-460: 577-613.
[30]Paatero J, Hamrie K, Jaakkola T, et al. Airborne and deposited radioactivity from the Chernobyl accident: a review of investigations in Finland[J]. Boreal Environment Research, 2010, 15: 19-33.
[31]Malá H, Rulík P, Cková V B, et al. Particle size distribution of radioactive aerosols after the Fukushima and the Chernobyl accidents[J]. J Environ Radioact, 2013, 126: 92-98.
[32]Bossew P, Kirchner G, de Cort M, et al. Radioactivity from Fukushima Daiichi in air over Europe; part 1: spatio-temporal analysis[J]. J Environ Radioact, 2012, 114: 22-34.
[33]Kirchner G, Bossew P, de Cort M. Radioactivity from Fukushima Daiichi in air over Europe; part 2: what can it tell us about the accident?[J]. J Environ Radioact, 2012, 114: 35-40.
[34]Lewis B J, Thompson W T, Iglesias F C. Invention in comprehensive nuclear materials[M]. Amsterdam: Elsevier, 2012, 2: 515-546.
[35]Kinoshita N, Suekia K, Sasa K, et al. Assessment of individual radionuclide distributions from the Fukushima nuclear accident covering central-east Japan[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108, 49: 19526-19529.
[36]Winiarek V, Bocquet M, Saunier O, et al. Estimation of errors in the inverse modeling of accidental release of atmospheric pollutant: application to the reconstruction of the cesium-137 and iodine-131 source terms from the Fukushima Daiichi power plant[J]. Journal of Geophysical Research, 2012, 117: D05122.
[37]Korsakissok I, Mathieu A, Didier D. Atmospheric dispersion and ground deposition induced by the Fukushima Nuclear Power Plant accident: a local-scale simulation and sensitivity study[J]. Atmos Environ, 2013, 70: 267-279.
[38]Chino M, Nakayama H, Nagai H, et al. Preliminary estimation of release amounts of131I and137Cs accidentally discharged from the Fukushima Daiichi Nuclear Power Plant into the atmosphere[J]. J Nucl Sci Technol, 2011, 48(7): 1129-1134.
[39]Masson O, Baeza A, Bieringer J, et al. Tracking of airborne radionuclides from the damaged Fukushima Daiichi nuclear reactors by European networks[J]. Environ Sci Technol, 2011, 45: 7670-7677.
[40]Morino Y, Ohara T, Nishizawa M. Atmospheric behavior, deposition, and budget of radioactive materials from the Fukushima Daiichi Nuclear Power Plant in March 2011[J]. Geophysical Research Letters, 2011, 38, L00G11.L: 11.
Development of Chemical Analysis for Radioactive Aerosols From Fukushima Daiichi Nuclear Accident
XIE Bo, HU Sheng, LONG Xing-gui
Institute of Nuclear Physics and Chemistry, China Academy of Engineering Physics, Mianyang 621999, China
The Fukushima Daiichi Nuclear Power Plant(FDNPP) accident was rated at the maximum level of 7 on the International Nuclear Event Scale by the international society. This paper reviews the development of chemical analysis work for radioactive aerosols released during the early stages of the accident in many countries after the FDNPP accident, and reveals these aerosol particles bearing radionuclides, such as Cs, Pu, Xe and I. In this paper, some physical-chemical characterization and dynamic behaviors of radioactive aerosols with a few points, including particles sampling and collection, particles analysis and characterization, particles transport and deposition behavior, and identical accident comparison have been conducted.
aerosols; radionuclide; nuclear accident; chemical analysis
2016-04-18;
2016-10-20
谢 波(1975—),男,湖北潜江人,博士研究生,副研究员,核燃料循环与材料专业,E-mail: caepxiebo@163.com
O615.45
A
0253-9950(2017)02-0129-09
10.7538/hhx.2017.39.02.0129
