超高效液相色谱串联质谱法同时测定茶叶中11种植物生长调节剂及吡虫啉、啶虫脒的残留
2017-04-14诸力王晨陈红平张颖彬周苏娟王国
诸力+王晨+陈红平+张颖彬+周苏娟+王国庆+刘新
摘要建立了超高效液相色谱串联质谱法同时快速测定茶叶中11种植物生长调节剂(PGRs)及吡虫啉、啶虫脒的方法。样品经乙腈甲酸溶液(99〖KG-3∶〖KG-51,V/V)均质提取,采用C18、强阴离子交换剂(SAX)、N丙基乙二胺(PSA)和无水MgSO4混合吸附剂分散萃取处理,以HSST3色谱柱分离,采用电喷雾(ESI)正负离子同时扫描和可编程多反应监测模式(SMRM)检测,基质匹配溶液外标法定量。6苄氨基嘌呤、多效唑、烯效唑、氯吡脲、甲哌啶、吡虫啉、啶虫脒在1~200μg/L和2,4二氯苯氧乙酸、对氯苯氧乙酸、赤霉素、吲哚乙酸、萘乙酸、吲哚丁酸在5~1000μg/L范围内线性良好(R2>0.99)。13种化合物加标回收率在73.1%~108.9%之间,RSD(n=6)值在0.6%~8.0%之间,方法检出限(LOD,S/N=3)和定量限(LOQ,S/N=10)分别为0.18~9.68μg/kg和0.61~32.26μg/kg。本方法简便、稳定、灵敏,能够满足实际检测需求。
关键词茶叶;植物生长调节剂;吡虫啉;啶虫脒;超高效液相色谱串联质谱;分散固相萃取
1引言
植物生长调节剂(Plantgrowthregulators,PGRs)是一类与植物激素具有相似生理和生物学效应的农药(来源包括人工合成及生物中提取),自20世纪30年代以来,因其效用高、用量小、残毒少等特点,广泛应用于果蔬、茶叶等农业生产领域,如赤霉素[1]、吲哚丁酸[2]、奈乙酸[3]等可通过改变茶树体内的生物合成、运转和分布,调节新梢生理过程,并促进茶树早发、多发,提高产量及品质。吡虫啉和啶虫脒属于烟碱类新型广谱杀虫剂,在茶园中应用也十分普遍[4,5]。然而随着植物生长调节剂和烟碱类农药用量增多及使用范围日益扩大,其安全性也备受关注。研究表明,2赤霉素可能增加肿瘤形成风险[6],吲哚乙酸可影响心肌功能[7],2,4二氯苯氧乙酸(2,4D)具有生殖毒性[8],多效唑存在致畸作用[9]。因此,欧盟、日本、中国等建立了PGRs和烟碱类农药相关限量标准,如欧盟规定2,4D在蓝莓、茶叶中的最高残留限量值(MRL)为0.1mg/kg;日本規定赤霉素在柠檬等果蔬中的MRL为0.2mg/kg;我国GB27632014中规定氯吡脲在葡萄中的MRL为0.05mg/kg,吡虫啉在茶叶中的MRL为0.5mg/kg[10]等。
目前,植物生长调节剂和烟碱类农药主要检测方法包括色谱法[11~13]、色谱质谱法[14~19]等,但绝大部分方法都需要经过传统固相萃取、浓缩、衍生等前处理步骤,存在操作程序繁琐、污染严重、抗干扰差等缺点。前处理作为关键技术,2会直接影响方法灵敏度和精密度。茶叶具有典型基质效应,一般方法很难直接适用,迄今尚未发现同时测定茶叶中多PGRs及烟碱类农药残留相关研究报道。本研究采用分散固相萃取前处理方法,省去浓缩、固相小柱萃取等步骤,建立了超高效液相色谱串联质谱法同时快速测定茶叶中11种植物生长调节剂及吡虫啉、啶虫脒的方法,本法灵敏、简便、高效,可满足茶叶质量监测需求。
2实验部分
2.1仪器与试剂
ABSciexTQ5500型串联三重四极杆质谱仪(美国Sciex公司);LC30A超高效液相色谱仪(日本岛津公司);MillQ去离子水发生器(美国Millipore公司);T10高速均质仪(德国IKA公司);高速冷冻离心机(上海卢湘仪离心机仪器有限公司)。
11种PGRs及2种烟碱类农药标准样品:6苄氨基嘌呤(6Benzylaminopurine,6BA)、多效唑(Paclobutrazol)、烯效唑(Uniconazole)、氯吡脲(Forchlorfenuron)、甲哌啶(Mepiquatchloride)、吡虫啉(Imidacloprid)、啶虫脒(Acetamiprid)、2,4二氯苯氧乙酸(2,4Dichlorophenoxyaceticacid,2,4D)、对氯苯氧乙酸(4Chlorophenoxyaceticacid,4CPA)、赤霉素(Gibberellicacid,GA3)、吲哚乙酸(Indole3aceticacid,IAA)、萘乙酸(1Naphthaleneaceticacid,1NAA)、吲哚丁酸(Indole3butyricacid,IBA)(纯度≥95.0%,德国Dr.Ehrenstorfer公司)。
甲醇、乙腈(色谱纯,TEDIA公司);甲酸铵(99.9%)、甲酸(95%)(德国Sigma公司);N丙基乙二胺(PSA)、强阴离子交换剂(SAX)、强阳离子交换剂(SCX,美国安捷伦科技有限公司);C18、石墨化碳黑(GCB)、无水MgSO4、弗罗里硅土(天津博纳艾尔杰公司)。
茶叶样品购于浙江省各大茶叶市场。
2.2色谱和质谱条件
WatersHSST3色谱柱(100mm×2.1mm,1.7μm);流动相A:1mmol/L甲酸铵水溶液,流动相B:1mmol/L甲酸铵甲醇溶液;梯度洗脱程序:0~1min,10%B;1~2min,10%~70%B;2~6min,70%~98%B;6~8min,98%B;8~8.1min,98%~10%B;8.1~12min,10%B。流速:0.25mL/min;进样量:1.0μL;柱温:40℃;运行时间:12min。
离子源:电喷雾离子源(ESI),温度500℃,电压5500V(ESI+)/ 4500V(ESI );雾化气(GS1)压力:334.7kPa;辅助气(GS2)压力:334.7kPa;气帘气(CurtianGas)压力:241.3kPa。定性与定量离子对、去簇电压(DP)、碰撞能量(CE)等参数见表1。
2.3样品制备
茶叶样品粉碎后过200μm筛,称取2.0g(精确至0.01g)试样于50mL离心管中,加入乙腈甲酸溶液(99〖KG-3∶〖KG-51,V/V)10mL,涡旋振荡1min,18000r/min均质提取2min,10000r/min低温离心10min(5℃),取上清液1mL至装有100mgC18,50mgSAX,25mgPSA和50mg无水MgSO4吸附剂的2mL离心管中,涡旋1min,10000r/min离心5min,上清液过0.22μm滤膜,待液相色谱串联质谱分析。
2.4标准曲线制作
分别称取13种化合物标准品各0.05g,用甲醇溶解并定容至50mL,分别配制成1000mg/L标准溶液,4℃保存,待用。
标准曲线的配制:茶叶空白样品按2.3节处理后,得到空白提取液,分别配制8个不同浓度(ESI+:1,2,5,10,20,50,100和200μg/L,ESI-:5,10,25,50,100,250,500和1000μg/L)混合基质标准溶液,现配现用。
3结果与讨论
3.1质谱和色谱条件优化
分别配制0.5mg/L浓度13种化合物溶剂标样进行质谱条件优化。采用ESI+和ESI-扫描模式,分别选择各准分子离子\[M+H\]+和\[M-H\]-为母离子,进行二级质谱优化,参数包括碰撞能量(CE)、去簇电压(DP)、电离温度等,优化结果详见表1。采用可编程多反应监测(Scheduledmultiplereactionmonitoring,SMRM),可以在不同时间段同时进行多种农药残留检测,较传统MRM相比,极大提高了单位检测通量,可有效改善峰型,提高精密度和重现性[20]。
使用HSST3、BEHC18和BEHC8色谱柱进行色谱条件优化,发现GA3、IAA等有机酸在HSST3柱上保留色谱峰型及分离度更佳。考察了包括甲醇水、乙腈水、甲醇水(0.1%甲酸和1mmol/L甲酸铵)、甲醇水(1mmol/L甲酸铵)在内的流动相。结果表明,采用甲醇水(0.1%甲酸和1mmol/L甲酸铵)作为流动相,虽然可提高\[M+H\]+化合物离子化效率,但甲酸中H+会降低\[M-H\]-化合物灵敏度。缓冲体系可进一步增加系统稳定性,从而使13种化合物分离度和灵敏度达到最佳,故流动相采用甲醇〖CM(44水(1mmol/L甲酸铵)。图1为空白茶样和加标茶样中13种化合物二级质谱总离子流色谱图,可见其〖CM)
分离效果良好,目标化合物出峰段杂质干扰较小。
3.2前处理条件优化
由于植物生長调剂种类繁多,化合物结构特性差异较大,通常采用乙腈、甲醇或其含甲酸[13,14,22](乙酸)溶液等进行提取。本研究考察了纯乙腈、乙腈水(80〖KG-3∶〖KG-520,V/V)、乙腈甲酸溶液(99〖KG-3∶〖KG-51,V/V)、乙腈/水/甲酸溶液(80〖KG-3∶〖KG-519〖KG-3∶〖KG-51,V/V)的提取效率。结果表明,采用乙腈水甲酸溶液(80〖KG-3∶〖KG-519〖KG-3∶〖KG-51,V/V)提取时,部分化合物回收率略优于乙腈甲酸溶液(99〖KG-3∶〖KG-51,V/V),但同时会对绝大部分化合物,尤其是GA3和4CPA,产生较强基质效应(Matrixeffect,Me),导致质谱信号严重降低,信噪比减小,影响其检出限。原因可能是使用含水的乙腈溶剂提取时,茶叶中强极性杂质共溶出,导致各化合物结构改变或离子化效率降低。在提取过程中添加适量甲酸可增加2,4D、4CPA等含羧基酸性PGRs提取效率,并且有效降低净化过程中此类化合物被吸附率[21],同时可防止GA3等在碱性环境中降解。因此本研究选择乙腈甲酸溶液(99〖KG-3∶〖KG-51,V/V)作为提取剂。
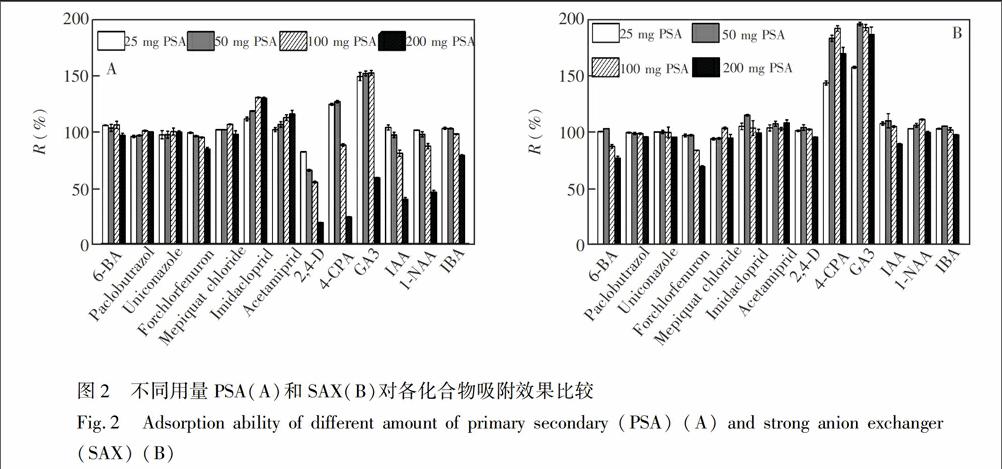
对于净化吸附剂选择,分别称取25、50、100和200mg的7种吸附剂(PSA、SAX、SCX、C18、GCB、无水MgSO4、弗罗里硅土),各加入1mL浓度为80μg/L(ESI+)、400(ESI-)μg/L基质混合标准溶液。净化效果用R值表示,R=〖SX(S〖S0〖SX)×100%,其中S0为净化前基质标样峰面积,S为净化后基质标样峰面积。R值越高,表示经净化后化合物信号越强,被吸附率越低,净化效果越理想。实验结果表明,C18和无水MgSO4吸附作用不明显,其它5种吸附剂对目标化合物吸附作用随用量增加而增强。其中SCX、GCB和弗罗里硅土对多种化合物有较强吸附作用,如25mgSCX吸附后,6BA和氯吡脲R值分别为0.7%和2.6%;50mgGCB吸附后6BA和4CPA的R值分别为18.6%和56.4%等。如图2所示,PSA和SAX在低用量时R值较高,随用量增加R值下降,25mgPSA和50mgSAX吸附后各化合物R值范围分别为82.3%~171.2%和93.5%~194.8%,净化效果良好。
SAX是一种以高纯球形硅胶键合季胺基团的吸附剂,常用于去除强阴离子杂质(如有机酸,无机离子等),PSA(N丙基乙二胺)为弱阴离子交换剂,可有效去除茶叶中色素、脂肪酸等杂质,但当两者用量提高时,对目标化合物吸附作用明显,如200mgPSA吸附后,2,4D的R值仅为23.8%,与文献\[22\]的结果相符。C18具有强疏水特性,对脂肪酸等弱极性化合物具有较强吸附作用。综合比较,本研究选择100mgC18、50mgSAX、25mgPSA和50mg无水MgSO4作为吸附剂,净化效果最佳。
3.3方法的线性范围、检出限和定量限
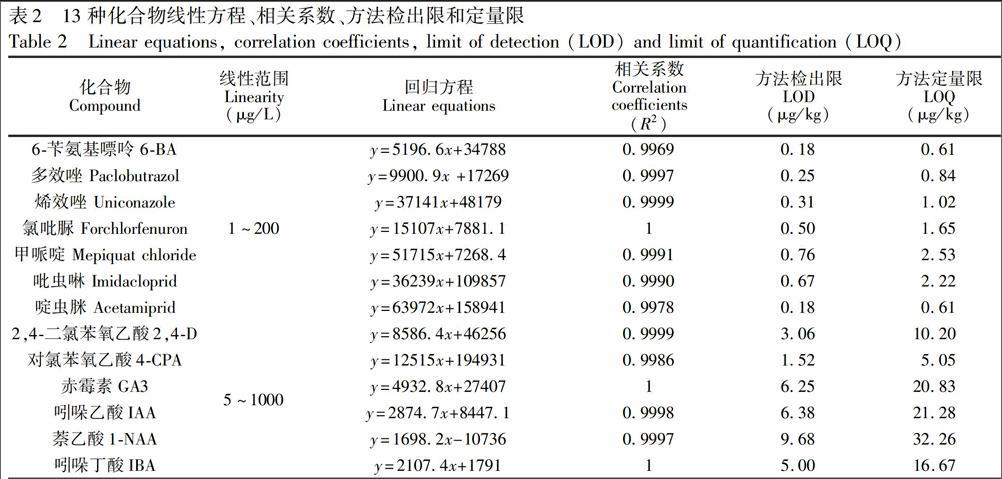
采用2.4节制备的各浓度基质混合标准溶液,按优化后参数测定,以各定量离子对峰面积对应浓度做标准曲线,得到13种化合物线性方程及其相关系数,如表2所示。6BA、多效唑、烯效唑、氯吡脲、甲哌啶、吡虫啉、啶虫脒在1~200μg/L和2,4D、4CPA、GA3、IAA、1NAA、IBA在5~1000μg/L范围内线性关系良好(R2>0.99)。方法检出限(LOD,S/N=3)和定量限(LOQ,S/N=10)分别为0.18~9.68μg/kg和0.61~32.26μg/kg。
3.4基质效应
根据相应浓度甲醇溶剂标准样品进行基质效应评价[23],计算公式为Me=〖SX(kmethanol-kmatrix〖kmethanol〖SX)×100%(k为标准曲线斜率)。结果表明,13种化合物基质效应范围在 49.5%~56.2%之间。为消除基质效应干扰,实验采用相应基质匹配标样进行校准。
3.5回收率和精密度
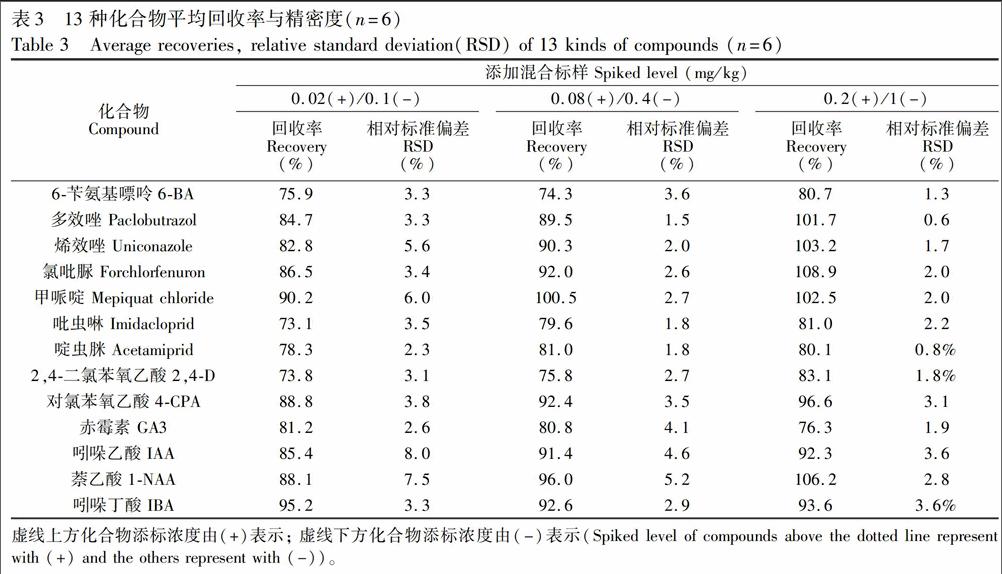
称取茶叶2.0g,分别添加0.02,0.08和0.2mg/kg(ESI+)和0.1,0.4和1.0mg/kg(ESI-)度的混合标准溶液,按照前述方法处理,平行测定6次,回收率和精密度数据见表3。由表3可见,13种化合物加标回收率范围在73.1%~108.9%之间,RSD(n=6)在0.6%~8.0%之间,表明本方法重复性良好。
虚线上方化合物添标浓度由(+)表示;虚线下方化合物添标浓度由(-)表示(Spikedlevelofcompoundsabovethedottedlinerepresentwith(+)andtheothersrepresentwith(-))。[BG)W][HT5][HJ]
3.6实际样品分析
采用本方法分析了市售69份茶叶样品,所有茶样均未检出PGRs,其中11份检出吡虫啉(0.023~0.612mg/kg,1份超过GB2763最大残留限量0.5mg/kg),13份检出啶虫脒(0.014~0.831mg/kg)。原因可能是测试茶样多为夏秋季采收,其经济价值较低,且病虫害严重,因此为降低成本,PGRs用量减少,而杀虫剂用量增加。
4结论
本研究建立了适用于茶叶中11种PGRs及吡虫啉、啶虫脒测定的超高效液相色谱串联质谱检测方法。本方法简便快捷,灵敏度高,重现性好,可满足欧盟、日本等国对茶叶中PGRs等农药的限量要求,为我国茶叶行业制定相关检测标准提供了理论依据。
References
1ChenHP,LiuX,YangD,YinP.FoodChem.,2013,138(2):976-981
2PaulA,LalL,AhujaPS,KumarS.Mol.Bio.Rep.,2012,39(11):3485-3490
3JalaA,HankamolsiriW.ActaHortic.,2014,1023:233-240
4ZhangYY,LuHY,WangB,ZhangZ,LinXR,ChenZZ,LiB.Int.J.FoodSci.Tech.,2015,50(6):1397-1404
5HouRY,JiaoWT,QianXS,WangXH,XiaoY,WanXC.J.Agr.FoodChem.,2013,61(51):12565-12571
6ErinN,AfacanB,ErsoyY.Toxicology,2008,254(12):75-81
7TuluceY,CelikI.AsianJ.Chem.,2006,18(1):528-532
8LeeKJ,JohnsonV,BlakleyB.Toxicology,2001,165(1):39-49
9WangKS,LuCY,ChangSH.J.Hazard.Mater.,2011,190:520-528
10GB27632014,NationalFoodSafetyStandardMaximumResidueLimitsforPesticidesinFood.NationalStandardsofthePeople′sRepublicofChina
食品中国家安全标准食品中农药最大残留限量.中华人民共和国国家标准.GB27632014
11VogelA.Bull.Environ.Contam.Toxicol.,1998,60(3):371-378
12WangMY,ChangXC,WuXY,YanHY,QiaoFX.J.Chromatogr.A,2016,1458:9-17
13LiGL,LiuSC,SunZW,XiaL,ChenG,YouJM.FoodChem.,2015,170:123-130
14WUPingGu,TANYing,ZHANGJing,WANGLiYuan,TANGYun,JIANGWei,PANXiaoDong,MABingJie,NIZhuNan,WANGTianJiao.ChineseJ.AnalChem.,2014,42(6):866-871
吳平谷,谭莹,张晶,王立媛,汤鋆,姜维,潘晓东,马冰洁,倪竹南,王天娇.分析化学,2014,42(6):866-871
15LIUJingJing,GONGPing,ZHANGXiaoMei,WANGJianHua,WANGJingTang.ChineseJournalofChromatography,2012,30(10):1012-1016
刘靖靖,宫萍,张晓梅,王建华,王境堂.色谱,2012,30(10):1012-1016
16MOUYanLi,GUODeHua,DINGZhuoPing,YIXiongHai.ChineseJ.Anal.Chem.,2013,41(11):1640-1646
牟艳莉,郭德华,丁卓平,伊雄海.分析化学,2013,41(11):1640-1646
17HauJ,RiedikerS,VargaN,StadlerRH.J.Chromatogr.A,2000,878(1):77-86
18CAIYiPing,SUNZhiWei,WANGXiaoYan,SUOYouRui,YOUJinMao.ChineseJ.Anal.Chem.,2015,43(3):419-423
蔡轶平,孙志伟,王小艳,索有瑞,尤进茂.分析化学,2015,43(3):419-423
19KimKG,ParkDW,KangGR,KimTS,YangYS,MoonSJ,ChoiEA,HaDR,KimES,ChoBS.FoodChem.,2016,208:239-244
20HangK,WongJW,YangP,HaywardDG,SakumaT,ZouYY,SchreiberA,BortonC,NguyenTV,KaushikB,OulkarD.Anal.Chem.,2012,84(13):5677-5684
21WiestL,BuleteA,GiroudB,FrattaC,AmicS,LambertO,PouliquenH,ArnaudguilhemC.J.Chromatogr.A,2011,1218(34):5743-5756
22JIANGZhenHui,ZHAWuLa,YINGTieJin.J.Chin.Inst.FoodSci.Tech.,2015,15(11):192-198
蔣振晖,扎乌拉,应铁进.中国食品学报,2015,15(11):192-198
23KittlausS,SchimankeJ,KempeG,SpeerK.J.Chromatogr.A,2011,1218(46):8399~8410
AbstractAnefficientmethodfortheanalysisofmulticlassplantgrowthregulatorsandpesticide(imidacloprid,acetamiprid)residuesinteawasdevelopedbasedonultraperformanceliquidchromatographytandemmassspectrometry(UPLCMS/MS).Thesampleswereextractedwithacetonitrile/formicacid(99〖KG-3∶〖KG-51,V/V)solution,cleanedupwithfoursorbentsincludingC18,stronganionexchanger(SAX),primarysecondaryamine(PSA)andanhydrousMgSO4.ThecompoundswereseparatedonaHSST3columnunderpositive/negativeelectrosprayionizationmode,detectedbyscheduledmultiplereactionmonitoring(SMRM),andquantifiedbymatrixmatchedexternalstandardcurves.Allpesticideresiduesshowedgoodlinearityintheconcentrationrangeof1-200μg/L(6benzylaminopurine,paclobutrazol,uniconazole,forchlorfenuron,mepiquatchloride,imidacloprid,acetamiprid)or5-1000μg/L(2,4dichlorophenoxyaceticacid,4chlorophenoxyaceticacid,indole3aceticacid,gibberellicacid,1naphthaleneaceticacid,indole3butyricacid),withcorrelationcoefficient(R2≥0.99).Limitsofdetection(LOD,S/N=3)andlimitsofquantitation(LOQ,S/N=10)were0.18-9.68μg/kgand0.61-32.26μg/kg,respectively.Inaddition,thespikedrecoveriesofteasampleswere73.1%-108.9%,andRSDswere0.6%-8.0%.Thismethodwasappliedtocommercialsamples,andallthedetectionswereconfirmedbyacquiringtransitionsforeachpesticideinthesamples.
KeywordsTea;Plantgrowthregulator;Imidacloprid;Acetamiprid;Ultraperformanceliquidchromatographytandemmassspectrometry;Dispersivesolidphaseextraction
(Received4November2016;accepted20February2017)