94例非综合征性耳聋患儿基因突变结果分析
2017-04-13肖彩霞陈亚秋刘爽王洪月丁怡冰
肖彩霞 陈亚秋 刘爽 王洪月 丁怡冰
天津市妇女儿童保健中心五官听力科(天津 300070)
·临床研究·
94例非综合征性耳聋患儿基因突变结果分析
肖彩霞 陈亚秋 刘爽 王洪月 丁怡冰
天津市妇女儿童保健中心五官听力科(天津 300070)
目的分析非综合征性听力障碍儿童耳聋基因突变情况,从分子水平探究该人群聋病的遗传病因和特点,为早期诊断、治疗及预防先天性和遗传性耳聋提供科学依据。方法采用基质辅助激光解吸电离飞行时间质谱(MALDI-TOF MS)技术,对天津市儿童听力障碍诊治中心诊断的94例非综合征性听力障碍儿童进行常见耳聋基因(GJB2、GJB3、SLC26A4、线粒体12SrRNA)共20个突变位点的检测,并对检测结果及听力学资料进行统计学分析。结果94例研究对象皆为中度、重度及极重度感音神经性耳聋,其中双侧听力下降组73例,单侧听力下降组21例。94例患儿中31例(32.98%,31/94)检出携带耳聋易感基因突变,单基因纯合突变13例,单基因复合杂合突变9例,单杂合突变9例,其中双侧听力下降组耳聋基因阳性率(42.47%)明显高于单侧听力下降组(0.00%)(χ2=13.31,P<0.01)。GJB2基因、SLC26A4基因、GJB3基因及12SrRNA的突变检出率分别为17.02%、15.96%、0.00%和0.00%。GJB2阳性例数16例,皆位于双侧听力下降组,其中纯合突变8例,复合杂合突变5例,杂合突变3例。SLC26A4(PDS)基因阳性例数15例,皆位于双侧听力下降组,其中纯合突变者5例(皆位于IVS7-2A>G位点),复合杂合突变者4例,杂合突变者6例。双耳听力下降组的GJB2基因阳性检出率(21.92%)高于单耳听力下降组(0.00%)(P<0.05);SLC26A4基因阳性检出率两组间比较无明显差别(20.55%,0.00%)(P>0.05)。结论遗传因素在非综合征性耳聋的致聋病因中所占比例较高,双侧聋遗传性高于单侧聋,对于双侧耳聋及耳聋基因阳性患儿定期进行听力学随访意义重大,耳聋基因筛查是对常规听力学筛查的有效补充,可为降低出生缺陷的三级预防措施提供理论和实践依据。
非综合征性聋;GJB2;SLC26A4;基因突变
先天性耳聋是最常见的出生缺陷之一,60%与遗传因素有关,以听力丧失为唯一症状的非综合征性耳聋(non-syndromic hearing impairment,NSHI)占所有遗传性聋的70%,除耳聋外还合并其他系统异常的综合征性耳聋占30%[1]。目前的研究认为GJB2、SLC26A4基因和线粒体 DNA l2SrRNA (m.1555A>G和m.1494C>T)突变是导致非综合征性耳聋(NSHI)的常见分子病因,该3个基因已作为临床耳聋基因的常规检查,但在不同民族和不同地区的耳聋人群中突变热点和发病率有明显差异[2]。既往我们通过对天津市新生儿进行4个基因(GJB2/ GJB3/SLC26A4/线粒体l2SrRNA)20个突变位点的耳聋基因普遍筛查,发现天津市新生儿耳聋基因携带率为5.52%[3],然而目前对天津市听力障碍儿童中耳聋基因携带情况未见报道。本研究通过对天津地区的94名非综合征性听力障碍儿童进行耳聋基因检测,以了解天津地区听力障碍儿童中耳聋基因携带情况,同时揭示本地区遗传性耳聋基因流行病学情况。
1 资料与方法
1.1临床资料
所有研究对象均为天津市新生儿听力筛查未通过或家长发现患儿对声音反应差转诊至天津市儿童听力障碍诊治中心,并经我中心诊断的非综合征性感音神经性耳聋患者,确诊听力障碍年龄3个月~3岁不等,共94例。检测年龄为2岁到16岁不等,平均(6.64±2.08)岁,其中男58例,女36例。
1.2听力学诊断及评估
回顾性分析患儿于我中心进行听力学诊断及随访的临床资料,包括患儿的一般信息,母孕期疾病史、耳毒性药物用药史,新生儿及生后疾病史、头部外伤史、耳聋家族遗传史、体格检查(全身及专科查体)资料、影像学(颞骨CT或MRI)资料、出生时听力筛查资料、听力学诊断及后期随访资料等部分组成。听力学诊断采用声导抗及电生理检查,具体如下:(1)声导抗测试:应用美国GSI-33型中耳分析仪中耳分析仪进行声导抗测试,1岁以下采用1 kHz及226Hz探测音。(2)电生理检查:采用美国GSI公司的Audera听觉脑干诱发电位测试仪进行测试,在隔音屏蔽室内按照常规方法行听性脑干反应测试。以短声ABRⅤ波反应阈值作为婴幼儿2~4 kHz高频听力损失的参考指标。以ABR波Ⅴ反应阈≤30 dBnHL作为听力正常的指标,听力损失程度分级标准[4]具体如下:轻度聋:波Ⅴ反应阈值为31~50dBnHL;中度聋:51~70 dBnHL;重度聋:71~90 dBnHL;极重度聋:≥91 dBnHL。
1.3基因检测方法
在知情同意情况下采集研究对象末梢循环血于滤纸片上,由华大基因检验所利用基质辅助激光解析离子飞行时间质谱技术检测与耳聋相关的4个基因中的20个突变位点,分别是GJB2基因(c.235delC,c.299_300delAT,c.167delT,c.176_ 191 del16,c.35delG)、SLC26A4(PDS)基因(c.1174A>T, c.1226G>A, c.1229C>T,c.1975G>C,c.2027T>A, c.2162C>T,c.2168A>G,c.281C>T,c.589G
>A,c.IVS15+5G>A,c.IVS7-2A>G)、GJB3基因(c.538C>T,c.547 G>A)、线 粒 体 12SrRNA基 因(m.1494C>T,m.1555A>G)。
2 统计学方法
应用SPSS 17.0软件,双侧听力下降组与单侧听力下降组基因突变阳性率的比较采用卡方检验,两组GJB2及SLC26A4基因阳性检出率采用校正卡方检验,以P<0.05为差异具有统计学意义。
3 结果
3.1耳聋分型
94例研究对象皆为中度、重度及极重度感音神经性耳聋,双侧听力下降组73例,单侧听力下降组21例。双耳对称性极重度耳聋45例,双耳对称性重度聋5例,双耳对称性中度聋5例,7例为一耳重度一耳极重度耳聋,余11例为一耳中度一耳重度或极重度耳聋。单耳极重度耳聋15例,重度聋4例,中度聋2例。
94例研究对象有耳聋遗传家族史者16例;单耳耳廓畸形、耳道闭锁者1例;人工耳蜗已植入者25例,佩戴助听器者57例。
3.2 耳聋基因检测结果
94例患儿中31例(32.98%,31/94)检出携带耳聋易感基因突变,其中单基因纯合突变13例,单基因复合杂合突变9例,单基因杂合突变突变9例;其中双侧听力下降组阳性检出率为42.47%(31/73),单侧听力下降组阳性率为0.00%(0/21),二者比较差异有显著的统计学意义(χ2=13.31,P=0.000)。
GJB2基因阳性例数16例(17.02%,16/94),双侧听力下降组GJB2阳性检出率为21.92%(16/73),单侧听力下降组检出率为0.00%(0/21),二者比较差异有统计学意义(校正χ2=4.10,P=0.04);其中GJB2纯合突变8例,复合杂合突变5例,杂合突变3例。
SLC26A4(PDS)基因阳性者15例(15.96%,15/ 94),皆位于双侧听力下降组,检出率为20.55%(15/ 73),单侧听力下降组检出率为0.00%(0/21),二者比较无统计学意义(校正χ2=3.72,P=0.054)(P>0.05);其中纯合突变5例(皆位于IVS7-2A>G位点),复合杂合突变者4例,杂合突变者6例。此次研究中GJB3及线粒体12SrRNA基因阳性者未检出。具体耳聋基因检出情况及听力分级见表1至表3。

表1 基因位点突变的检出情况[例(%)]Table 1 Detection of gene mutations[Cases(%)]
3.3 SLC26A4基因与影像学结果相互验证
94例研究对象中影像学提示存在大前庭导水管扩张者为16例,而其中SLC26A4基因阳性者有14例,大前庭导水管综合征患儿中SLC26A4基因阳性检出率为87.5%(14/16),其中5例为SLC26A4纯合突变,4例为复合杂合突变,5例为杂合突变。另外有两例患儿耳聋基因检测结果呈阴性。
15例SLC26A4基因阳性患儿中14例影像学提示大前庭水管扩张,纯合突变及复合杂合突变皆存在大前庭水管扩张,6例杂合突变者有中5例有大前庭导水管扩张,1例影像学未见异常。如图1
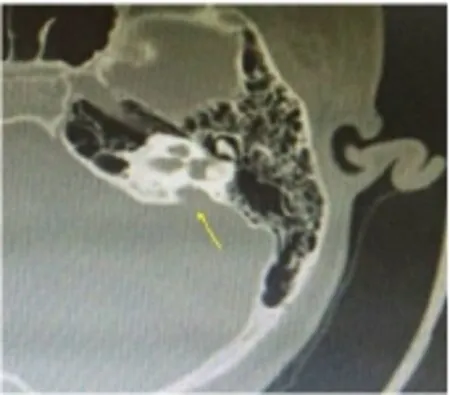
图1 大前庭导水管综合征患者颞骨CT结果(黄色箭头示扩大的前庭水管)Fig.1 CT scan of a case with large vestibu1ar aqueduct syndrome(The yellow arrow pointing large vestibu1ar aqueduct)
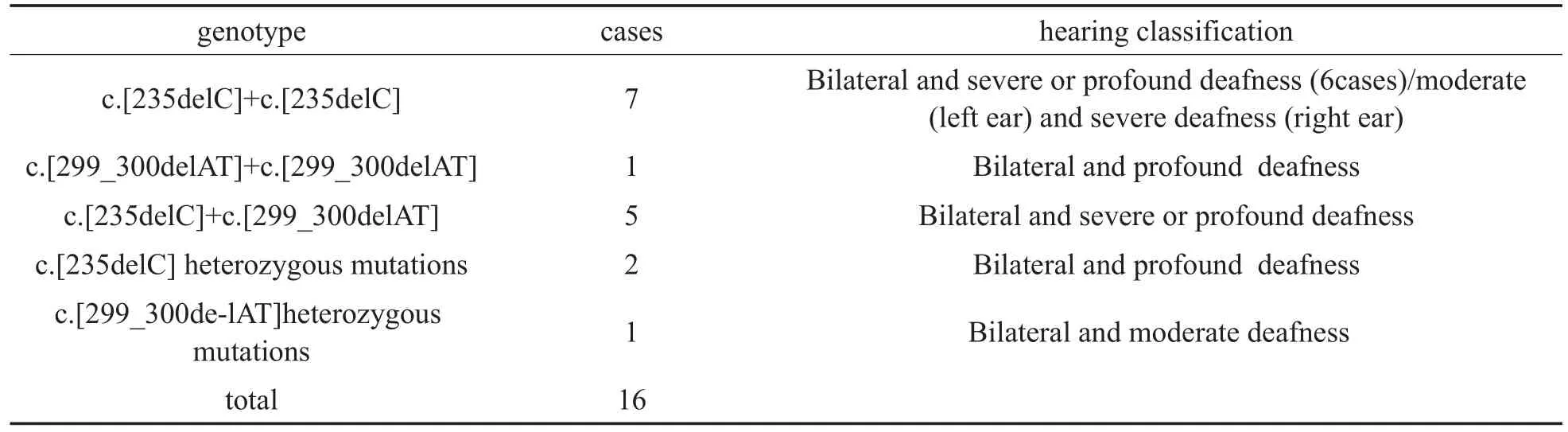
表2 GJB2基因阳性及听力分级情况Table 2 The positive cases of GJB2 gene and hearing classification
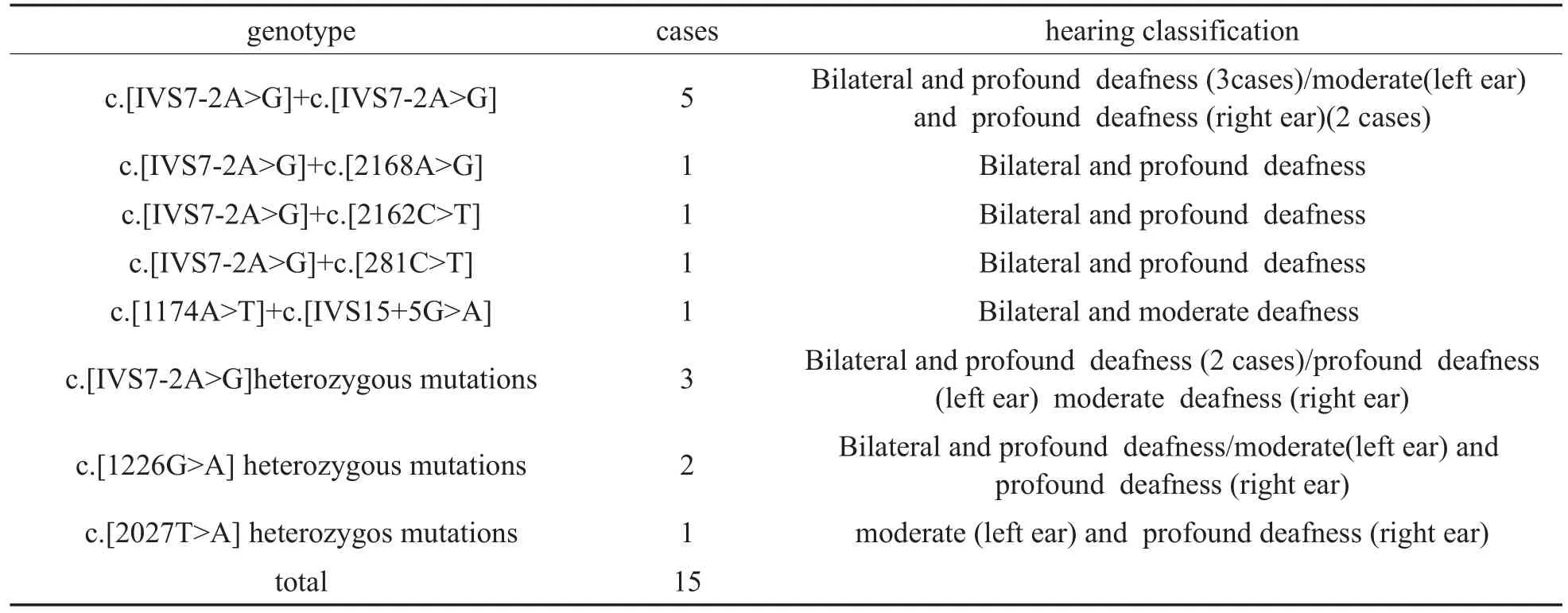
表3 SLC26A4(PDS)基因阳性及听力分级情况Table 3 The positive cases of SLC26A4(PDS)gene and hearing classification
3.4出生时听力筛查及随访情况
73例双侧耳聋组中有2例患儿出生时听力通过,余均未通过。2例均为SLC26A4(PDS)基因复合杂合突变者,待1岁左右患儿对声音反应差,行ABR检测提示为一例为双耳极重度聋,一例为双耳中度聋,颞骨CT均提示大前庭导水管扩张。一例单侧耳聋患儿(右侧耳廓畸形、耳道闭锁),6个月听力学诊断为右耳极重度耳聋,行MRI检查提示左耳未见明显异常,1岁时外伤后左耳听力极度下降,再次行ABR检测提示双耳极重度耳聋,颞骨CT提示左耳大前庭导水管扩张,而本次耳聋基因检测未检出阳性突变。
4 讨论
既往天津地区对58397名新生儿GJB2、GJB3、SLC26A4、12SrRNA 4个基因20个位点筛查研究中发现新生儿中耳聋基因携带率为5.52%,由GJB2和SLC26A4两种基因引起的听力损失占到0.58%[3]。本研究对94例存在听力障碍的非综合征性感音神经性耳聋患儿进行耳聋基因检测发现听力障碍儿童中耳聋基因携带率达32.98%,双侧耳聋患儿中耳聋基因阳性率则高达42.47%(31/ 73),远远高于本市新生儿中耳聋基因阳性率(5.52%)[3]。本研究中还发现双侧听力下降组患儿耳聋基因阳性检出率(42.47%)明显高于单侧听力下降组(0.00%)(P<0.01),其中GJB2基因阳性检出率(21.92%)高于单侧听力下降组(0.00%)(P<0.05),这与国内学者王国建等[5]报道一致,此结果提示GJB2及SLC26A4这两种基因突变多导致双侧耳聋,这一结果符合遗传性疾病的表型对称性特点。双侧听力下降组SLC26A4基因阳性检出率虽高于单侧听力下降组,然而与王国建等[5]报道不同两组比较无明显差别(P>0.05),可能与单侧听力下降组样本量较少有关。单侧听力下降组中未检出阳性突变,提示单侧耳聋的病因更多的可能为环境因素所致,也就是说双侧耳聋者遗传性要高于单侧耳聋者。因此,在日常工作中,对于先天性耳聋患者,尤其是双侧耳聋患者,要尤其注意耳聋基因病理性突变的问题,这对探究耳聋的病因至关重要。
GJB2基因是引起非综合征性耳聋的最常见的耳聋基因,本研究中检出的GJB2基因突变居第一位,占到基因阳性人数的51.6%(16/31),双耳听力下降组中GJB2检出率达21.92%(16/73),与以往文献报道一致[5,6]。GJB2常表现为常染色体隐性遗传,此研究中有13例患儿为GJB2纯合突变及复合杂合突变,此突变为该聋儿的致病性突变,听力学表现为中度、重度及其以上耳聋,提示GJB2纯合突变及复合杂合突变所致的耳聋,程度多较重,如果不早期干预可严重影响患儿言语发育[7]。然而3例GJB2杂合突变者也出现耳聋,虽耳聋基因检测为杂合突变,仍提示可能为遗传性耳聋,推测患儿、其父/母亲可能带有其它未知或罕见的致聋突变基因,有待进一步做GJB2基因的全序列检测,明确耳聋病因,这对聋儿的治疗和预后有重要意义[8]。
本研究中检出SLC26A4(PDS)基因阳性者15例,双侧耳聋中PDS阳性率为20.55%(15/73),这与天津市新生儿中PDS基因检出率位于第二位报道相同[3]。既往研究报道约在80%的大前庭水管患者中可发现SLC26A4突变,1%的正常人携带此种杂合突变[2]。本研究中发现16例影像学提示大前庭水管扩张患者中SLC26A4基因阳性率高达87.5% (14/16),其中5例纯合突变及4例复合杂合突变者均为大前庭导水管扩张,6例杂合突变者有5例有大前庭导水管扩张,1例影像学检查未见异常,此结果提示SLC26A4基因纯合及复合杂合突变患者均会发病,筛查结果为杂合突变者也有可能发病,对于此类患者也应积极地行该基因的全序列检测以明确致病原因,为遗传咨询指导提供重要的理论基础,避免再次生育聋儿。
73例双侧耳聋组中有2例患儿出生时听力通过,皆为SLC26A4复合杂合突变者,待1岁左右患儿对声音反应差,行听力学诊断提示为一患儿为双耳极重度聋,一患儿为双耳中度聋,影像学均提示大前庭导水管扩张。本研究提示耳聋基因筛查可以较早发现迟发性耳聋患儿,早期大前庭导水管综合征患者可以听力正常或轻度异常,头部外伤、重感冒、气压性创伤或其他使颅内压增高的诱因均有可能造成患者听力下降,因此对于携带SLC26A4基因的患者我们应定期进行听力学随访,并通过积极的临床指导延缓患者发病时间,保护患者的残存听力。同时也提示新生儿耳聋基因筛查是对于听力筛查的有效补充。一例单耳耳廓畸形、耳道闭锁患儿,6个月ABR检测诊断为单耳极重度耳聋,行MRI检查提示右耳听骨链完整,耳蜗螺旋一周,内听道狭窄、蜗神经缺如,左耳未见异常,1岁时因外伤后健耳听力极度下降,再次行听力学诊断提示双耳极重度耳聋,行颞骨CT提示之前健耳大前庭导水管扩张。因大前庭导水管疾病的特殊性,大前庭导水管或内淋巴管、内淋巴囊未扩张到一定程度时影像学检查可能呈阴性,因此对于临床表现怀疑为前庭导水管扩张患儿可再次行影像学检查,以免误诊,耽误治疗时机。
本研究中未检出线粒体DNA12SrRNA基因及GJB3基因常见致聋突变,主要是于本研究对象例数少有关。
5 小结
遗传因素在非综合征性耳聋的致聋病因中所占比例较高,双侧聋遗传性高于单侧聋,对于双侧及基因阳性患儿定期进行听力学随访意义重大;在新生儿中开展致聋基因的筛查是对常规听力学筛查的有效补充,尤其是迟发性耳聋患儿能较早检出,真正实现早发现、早诊断、早干预,预防和减少耳聋残疾的发生,并为具有聋病易感基因的听力损失儿童家庭提供针对性的婚前、孕前、产前的遗传咨询指导,避免再次生育聋儿。
1 黄选兆,汪吉宝,孔维佳,等.实用耳鼻咽喉头颈外科学[M].2版.北京:人民卫生出版社,2010:976. Huang XZ,Wang JB,Kong WJ,et al.Practice of Otorhinolaryngolo⁃gy-Head and Neck Surgery[M].2nd.Beijing:People's Medical Publishing House,2010:976.
2 Bayazit YA,Yilmaz M.An overview of hereditary hearing loss[J]. ORL J Otorhinolaryngol Relat Spec,2006,68(2):57-63.
3 Zhang J,Wang P,Han B,et al.Newborn hearing concurrent genet⁃ic screening for hearing impairment-a clinical practice in 58,397 neonates in Tianjin,China[J].International Journal of Pediatric Otorhinolaryngology,2013,77(12):1929-1935.
4 张宇,王幼勤,郭洪源,等.听力学表型在大前庭水管综合征早期诊断中的意义[J].中华耳科学杂,2016,14(1):57-60. Zhang Y,Wang YQ,Guo HY,et al.Significance of audiological phe⁃notypes to early diagnosis of large vestibular aqueduct syndrome [J].Chinese Journal of Otology,2016,14(1):57-60.
5 王国建,袁永一,李荣,等.不同听力学表型人群中常见耳聋基因突变检出率的分析.[J].临床耳鼻咽喉头颈外科杂志,2011,25 (10):445-448. Wang GJ,Yuan YY,Li R,et al.Analysis of positive rate of common genetic mutations in 1448 cases with different hearing phenotype [J].Journal of Clinical Otorhinolaryngol Head and Neck Surgery, 2011,25(10):445-448.
6 张强伟,张芩娜,杨向茹.山西太原129例重度非综合征型感音神经性耳聋基因的筛查[J].中华耳科学杂志,2012,10(2) 224-226. Zhang QW,Zhang QN,Yang XR.Screening of common deafness genes in129 severe to profound non-syndromic sensorineural hearing loss patients in Taiyuan[J].Chinese Journal of Otology, 2012,10(2)224-226.
7 陶峥,马衍,欧阳治国,等.205例先天性非综合征型聋患儿GJB2基因突变分析[J].听力学及言语疾病杂志,2010,18(1): 67-68. Tao Z,Ma Y,Ouyang ZG,et al.Mutation in the GJB 2 gene in 205 children with congenital non-syndromic hearing impairment[J]. Journal of Audiology and Speech Pathology,2010,18(1):67-68.
8 贾婧杰,袁永一,戴朴,等.山东省滨州市特教学校耳聋学生分子病因学分析—GJB2 235delC突变、线粒体DNA12S rRN⁃AA1555G突变和SLC 26A4 IVS 7-2A>G突变筛查报告[J].中华耳科学杂志,2010,8(4):407-410. Jia JJ,Yuan YY,Dai P,et al.Molecular etiology analysis among students with profound hearing loss in a special education school in Shandong[J].Chinese Journal of Otology,2010,8(4):407-410.
Mutation analysis of the common deafness genes in 94 children with non-syndromic hearing impairment
XIAO Caixia,CHEN Yaqiu,LIU Shuang,WANG Hongyue DING Yibing
Tianjin Maternal and Child Health Care Center,Tianjin 300070 Corresponding author:CHEN yaqiu Email:qianyi3@sina.com
Objective To analyze the deafness gene mutations in children with non-syndromic hearing impairment (NSHI),and to investigate the genetic etiology and features of deafness disorders at the molecular level.Methods 94 children with NSHI diagnosed by The Center of Diagnosis and Treatment of Children’s Hearing Disorders in Tianjin were screened for 20 hotspot of hearing loss-associated mutations from GJB2,GJB3,SLC26A4 and mitochondrial 12S rRNA(MTRNR1)using the matrix-assisted laser desorption ionization-time of flight mass spectrometry(MALDI-TOF MS).The data of genetic screening was analyzed with correlation to audiologic testing results.Results In 94 cases with moderate to profound sensorineural hearing loss,73 of which presented with bilateral sensorineural hearing loss,and 21 with unilateral hearing loss.Deafness gene mutations were found in 31 subjects(32.98%,31/94),13 of which with single gene homozygous mutations,9 of which with single gene and double-site heterozygous mutations,and 9 with single-gene and single-site mutations.The positive detective rate of deafness gene mutations in bilateral hearing loss group was 42.47%but no deafness gene was detected in unilateral hearing loss group.The positive detective rate of GJB2 and SLC26A4 in 94 subjects were 17.02%and 15.96%respectively,and no mutations found on GJB3 and MTRNR1.Sixteen subjects showed GJB2 mutations,8 of which with homozygous mutations,5 with double-site heterozygous mutations, and 3 with single-site heterozygous mutations.Fifteen subjects with bilateral hearing loss were SLC26A4(PDS)-positive,5 of which were homozygous(at IVS7-2A>G),4 subjects with double-site heterozygous mutations,and 6 subjects with single-site heterozygous mutations.The positive detective rates for GJB2 and SLC26A4 genes in the bilateral hearing loss group are 21.92%and 20.55%respectively,however these genes were not detected in the unilateral hearing loss group.Conclusions High proportion of etiologies in patients with NSHI were due to genetic factors.The heritability of bilateral deafness was higher than that of unilateral hearing loss.Regularly audiology follow-up for bilateral deafness and deafness gene-positive children was of great significance.The union screening could provide theoretical and practical basis for the three level preventive measures to reduce birth defects.
non-syndromic hearing impairment;GJB2;SLC26A4;gene
R764
A
1672-2922(2017)01-51-6
2016-06-30审核人:王国建)
10.3969/j.issn.1672-2922.2017.01.011
肖彩霞,研究生,医师,研究方向:小儿听力学
陈亚秋,Email:qianyi3@sina.com
