超高效液相色谱-串联质谱法测定螺旋藻多糖的单糖组成
2017-04-10粟有志张菁楠
赵 丹, 冯 峰, 粟有志, 张菁楠, 于 莲, 苏 瑾*, 张 峰*
(1. 佳木斯大学药学院, 黑龙江 佳木斯 154000; 2. 中国检验检疫科学研究院,食品安全研究所, 北京 100176; 3. 伊犁出入境检验检疫局, 新疆 伊宁 835000)
研究论文
超高效液相色谱-串联质谱法测定螺旋藻多糖的单糖组成
赵 丹1,2, 冯 峰2, 粟有志3, 张菁楠1, 于 莲1, 苏 瑾1*, 张 峰2*
(1. 佳木斯大学药学院, 黑龙江 佳木斯 154000; 2. 中国检验检疫科学研究院,食品安全研究所, 北京 100176; 3. 伊犁出入境检验检疫局, 新疆 伊宁 835000)
建立了同时测定螺旋藻多糖水解产物中鼠李糖、木糖、阿拉伯糖、果糖、甘露糖、葡萄糖、半乳糖、甘露醇、核糖、岩藻糖、葡萄糖醛酸、半乳糖醛酸12种糖类化合物的超高效液相色谱-串联质谱分析方法。螺旋藻样品经超声波辅助提取,用三氟乙酸水解,经Waters Acquity BEH Amide色谱柱(100 mm×2.1 mm, 1.7 μm)分离,以10 mmol/L甲酸铵和10 mmol/L甲酸铵-乙腈为流动相,在电喷雾电离源负离子(ESI-)模式下,用多反应监测(MRM)模式检测。结果表明,12种糖类化合物的定量限为0.005~0.15 mg/kg,线性范围为0.05~5 mg/L。按照样品中每种糖本底含量的50%、100%、150%进行添加,回收率为80.21%~121.6%。应用该方法对螺旋藻样品进行分析,结果发现:大部分样品都能检测到岩藻糖、半乳糖、阿拉伯糖、鼠李糖、葡萄糖、果糖、木糖、核糖,含量在0.3~889.4 mg/g之间。此外,测定的15个样品中岩藻糖、半乳糖、阿拉伯糖、鼠李糖、葡萄糖、果糖、木糖、核糖是共有组分,含量差异较大,但在所有样品中均未检测到甘露醇和甘露糖。该方法的建立可为阐明螺旋藻多糖的结构组成及其活性提供技术支撑及基础数据。
超高效液相色谱-串联质谱;单糖;多糖;螺旋藻;质量评价
多糖是生物体内重要的生物大分子。近年来的研究[1,2]表明,多糖有特殊的生物活性,能提高机体的免疫功能,在预防医学、临床医学、保健食品等方面发挥重大作用。人们对多糖的早期研究多集中于药用植物类,如黄芪、灵芝多糖等的活性研究。随着海洋药物的兴起,对螺旋藻多糖的活性研究已成为目前研究的重点。测定螺旋藻多糖的单糖组成对其活性分析和质量控制具有重要意义。
目前,对多糖的测定方法有:硅胶薄层层析法[3],通过苯酚-硫酸、蒽酮-硫酸等显色反应的分光光度法[4-6],衍生化的气相色谱[7]或液相色谱-紫外检测法[8-10],液相色谱-蒸发光检测或示差荧光检测法[11,12],离子色谱-电化学检测法[13-15]等。上述方法尽管可以粗略测定多糖的总含量,但用于测定多糖中单糖组成则存在准确度低、分离度差、操作繁琐等缺点。近年来,质谱技术的发展为糖类化合物的分析提供了一个强有力的分析手段。王川丕等[16]建立了利用超高效液相色谱-质谱联用技术测定茶叶中8种单糖和寡糖的方法。然而,该方法检测的糖的种类偏少,此外,方法的基质也仅限于茶叶。本文建立了超高效液相色谱-串联质谱测定螺旋藻多糖中12种单糖组成的检测方法,成功应用于市售实际螺旋藻产品的单糖组分分析。
1 实验部分
1.1 仪器与试剂
鼠李糖(rhamnose)、木糖(xylose)、阿拉伯糖(arabinose)、果糖(fructose)、甘露糖(mannose)、葡萄糖(glucose)、半乳糖(galactose)、甘露醇(mannitol)、核糖(ribose)、岩藻糖(fucose)(纯度>95%,德国Dr. Ehrenstorfer公司);葡萄糖醛酸(glucuronic acid)、半乳糖醛酸(galacturonic acid)(纯度>95%,美国U. S. Pharma-copoeia公司);乙腈、甲醇(色谱纯,美国Fisher Scientific公司)、甲酸铵(色谱纯,中国Aladdin公司);三氟乙酸(TFA,纯度>99%,中国J&K Scientific公司)。
AB 5500型三重四极杆质谱,配有ESI源(美国AB公司); Acquity超高效液相色谱(美国Waters公司); Milli-Q去离子水发生器(美国Millipore公司); KQ-500DE型超声波清洗仪(昆山市超声仪器有限公司); DKN612C型烘箱(日本雅玛拓公司); AllegraTMX-22R型离心机(美国贝克曼公司)。XBridge Amide色谱柱(150 mm×4.6 mm, 3.5 μm)、Acquity BEH HILIC色谱柱(50 mm×2.1 mm, 1.7 μm)和Acquity BEH Amide色谱柱(100 mm×2.1 mm, 1.7 μm)(美国Waters公司), Inertsustain NH2色谱柱(100 mm×2.1 mm, 3 μm,日本岛津公司)。
1.2 标准溶液的制备
准确称取100 mg标准品(精确至0.1 mg),用乙腈-水(1∶1, v/v)配制成质量浓度为1 000 mg/L的标准储备液,储存于4 ℃冰箱中备用。并用乙腈-水(1∶1, v/v)稀释成质量浓度为10 mg/L的标准溶液,用于质谱条件优化。
移取1 000 mg/L的标准储备液,添加适量乙腈-水(1∶1, v/v),稀释成系列质量浓度(0.05~5 mg/L)的标准工作溶液。
1.3 样品处理
称取0.5 g的螺旋藻粉末,加12.5 mL水,于40 ℃水中以400 W超声提取40 min,离心。取上清液,加水超声离心过程重复2次,合并上清液,定容到100 mL。取2 mL定容后的溶液加入Sevage溶液(正丁醇-氯仿,1∶5, v/v)萃取多次以除去蛋白质,直至两相间无乳白色絮状物。取上清液,按照1∶1的体积比加入2 mol/L三氟乙酸溶液,放入120 ℃恒温干燥箱中水解3 h,水解液加入甲醇洗涤后氮吹,甲醇洗涤和氮吹过程重复3次,将三氟乙酸除去,用1 mL水复溶后再经0.22 μm水相滤膜过滤,上机分析。
1.4 仪器条件
1.4.1 色谱条件
色谱柱:Acquity BEH Amide柱;流动相:10 mmol/L甲酸铵溶液(流动相A)和10 mmol/L甲酸铵-乙腈溶液(流动相B);流速:0.2 mL/min;进样量:5 μL;柱温:30 ℃。梯度洗脱程序:0~7.50 min, 90%B~80%B; 7.50~9.50 min, 80%B~60%B; 9.50~9.51 min, 60%B~90%B; 9.51~13.00 min, 90%B。
1.4.2 质谱条件
离子源:ESI-;离子源温度:500 ℃;扫描方式:负离子模式;雾化气GS1压力:380 kPa;辅助气GS2压力:414 kPa;毛细管喷雾电压:-5.5 kV;入口电压:-10 V;出口电压:-13 V。12种糖的质谱分析参数见表1。
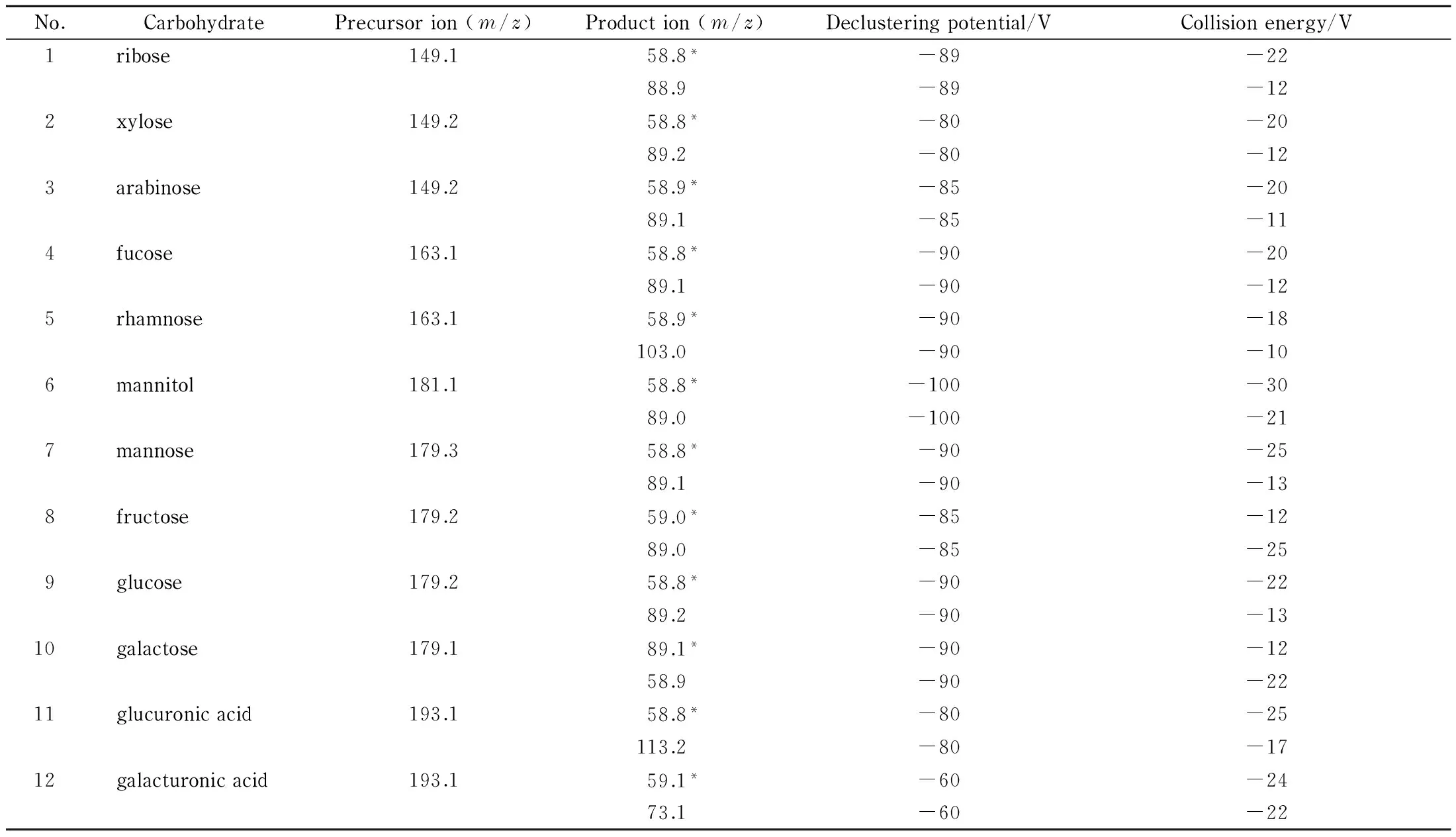
表 1 12种糖的监测离子对、去簇电压和碰撞气能量
* Quantitative ion.
2 结果与讨论
2.1 超声辅助提取多糖方法的优化
已有研究[17,18]表明,对于分析物中多糖的提取,超声辅助提取法的效果要优于常规加热提取法。为此,本研究重点对超声辅助提取方法中的超声时间、超声功率、提取温度、提取次数、液料比5个影响因素进行了优化,测定了岩藻糖、半乳糖、阿拉伯糖、鼠李糖、葡萄糖、果糖、木糖、核糖8种单糖的总糖提取率。结果如图1所示,最佳提取条件为提取2次,超声功率为400 W,提取温度为40 ℃,超声时间为40 min,液料比(体积质量比)为25 mL/g。

图 1 (a)提取次数、(b)超声功率、(c)提取温度、(d)超声时间、 (e)液料比对螺旋藻多糖中单糖提取率的影响(n=3) Fig. 1 Effects of (a) extraction times, (b) ultrasonic power, (c) extraction temperature, (d) ultrasonic time, (e) water/material ratio on the extraction efficiency of monosaccharides from spirulina polysaccharides (n=3)
2.2 色谱条件的优化
2.2.1 色谱柱的选择
单糖或寡糖富含羟基,属于高亲水的化合物,并且由于羟基的位点不同而有许多同分异构体。本研究中拟检测的12种单糖包含4组同分异构体:组1包括甘露糖、半乳糖、葡萄糖、果糖;组2包括岩藻糖、鼠李糖;组3包括阿拉伯糖、木糖、核糖;组4包括葡萄糖醛酸和半乳糖醛酸。比较了XBridge Amide柱、Acquity BEH HILIC柱、Inertsustain NH2柱和Acquity BEH Amide柱4款亲水色谱柱对12种糖的分离效果。以10 mmol/L甲酸铵-乙腈作为流动相,每种色谱柱在最优条件下对化合物进行分离。以组1为例,结果如图2所示,使用Acquity BEH HILIC柱和Inertsustain NH2柱时,4种同分异构体均没有完全分开;使用XBridge Amide柱与Acquity BEH Amide柱时,4种同分异构体都得到了分离,但使用XBridge Amide柱时,峰形较差。故选择Acquity BEH Amide柱作为分析柱。

图 2 半乳糖、甘露糖、葡萄糖、果糖在不同色谱柱中的总离子流图Fig. 2 Total ion chromatograms of galactose, mannose,glucose and fructose with different chromatographic columns a. XBridge Amide column; b. Acquity BEH HILIC column; c. Inertsustain NH2 column; d. Acquity BEH Amide column. Peaks: 1. mannose; 2. glucose; 3. fructose; 4. galactose.
2.2.2 流动相添加剂的选择
液相色谱-质谱法分析样品时,在流动相中添加适量的缓冲盐可调节流动相的pH值,提高分析的灵敏度,改善峰型,提高分离度。由于使用Acquity BEH Amide柱时,12种糖类化合物得到了分离,但未达到基线分离,因此考虑在流动相中添加适量的缓冲盐。本试验考察了10 mmol/L甲酸铵-10 mmol甲酸铵乙腈、0.1%(体积分数,下同)氨水-0.1%氨水乙腈、10 mol/L乙酸铵-10 mmol乙酸铵3种流动相对分离的影响。结果如图3所示,以组1为例,3种添加剂对4种糖的分离效果影响较大。添加剂为氨水时,甘露糖和果糖没有得到很好的分离,且出峰较晚;添加剂为乙酸铵时,只出现3个峰,且峰较宽;添加剂为甲酸铵时,4种糖得到了分离,虽然果糖、葡萄糖未达到基线分离,但不影响定量。流动相中添加氨水和乙酸铵并未改善分离度。因此,本试验选择甲酸铵作为流动相的添加剂。

图 3 半乳糖、甘露糖、葡萄糖、果糖在不同流动相中的总离子流图Fig. 3 Total ion chromatograms of galactose, mannose, glucose and fructose in different mobile phases a. 10 mmol/L ammonium formate-10 mmol/L ammonium formate/acetonitrile solution; b. 0.1% (v/v) ammonia 0.1% (v/v) ammonia/acetonitrile solution; c. 10 mmol/L ammonium 10 mmol/L ammonium acetate/acetonitrile acetate solution. Peaks: 1. mannose; 2. glucose; 3. fructose; 4. galactose.
2.3 质谱条件的优化
取10 mg/L标准溶液,通过针泵进样,在负离子检测方式下进行母离子全扫描,得到葡萄糖、半乳糖、木糖、甘露糖等的分子离子峰,以每种糖的分子离子峰为母离子,进行二级质谱扫描,采集全扫描的二级质谱图,得到碎片离子信息,再对每种糖的二级质谱参数进行优化,使每种糖的定性离子与定量离子产生的离子对强度达到最大时为最佳,得到最佳质谱参数(见表1)。经过色谱与质谱条件的优化,得到12种糖的MRM色谱图(见图4)。

图 4 12种糖的MRM色谱图Fig. 4 MRM chromatograms of 12 carbohydrates Peaks: 1. ribose; 2. xylose; 3. arabinose; 4. fucose; 5. rhamnose; 6. mannitol; 7. mannose; 8. glucose; 9. fructose; 10. galactose; 11. glucuronic acid; 12. galacturonic acid.
2.4 方法验证
2.4.1 线性关系、检出限、定量限
在优化好的色谱条件下,对12种糖类化合物在0.05~5 mg/L范围内进行了考察,分别以信噪比(S/N)为3和10时的质量浓度为检出限和定量限。阿拉伯糖、岩藻糖、鼠李糖、甘露糖、葡萄糖、果糖、半乳糖的线性范围为0.1~5 mg/L,核糖、木糖、甘露醇、葡萄糖醛酸、半乳糖醛酸的线性范围为0.05~2 mg/L。12种糖的检出限为0.002~0.05 mg/L,定量限为0.005~0.15 mg/kg(见表2)。
2.4.2 回收率和精密度
称取0.5 g螺旋藻样品,加入12种糖的混合标准溶液。通过对实际样品的测定,样品中含有岩藻糖、半乳糖、阿拉伯糖、鼠李糖、葡萄糖、果糖、木糖、核糖、葡萄糖醛酸和半乳糖醛酸10种单糖,未找到阴性样品,因此,按照样品中每种糖本底含量的50%、100%、150% 3个水平添加标准溶液,螺旋藻样品中不含有的甘露醇和甘露糖添加水平为10、20、30 μg/kg。按照样品处理方法制备后进行测定。加标处理后的样品溶液在一天内重复测定5次并计算RSD,即日内精密度;重复测定5天并计算RSD,即日间精密度。平均加标回收率为80.21%~121.6%,日内精密度和日间精密度分别为1.3%~4.8%和1.2%~5.0%(见表3),表明方法准确度好,精密度高,可对螺旋藻样品进行测定。
2.5 实际样品的检测
应用建立的UPLC-MS/MS方法对市售的螺旋藻样品进行了分析(见表4)。结果表明,所有螺旋藻样品均含有岩藻糖、鼠李糖、阿拉伯糖、半乳糖、葡萄糖、木糖、果糖、核糖8种单糖成分,这些单糖的含量在0.3~889.4 mg/g之间,另外在个别样品中检出了葡萄糖醛酸和半乳糖醛酸,它们的含量在5.1~420.4 mg/g之间。多种单糖在不同样品中含量差异较大,例如,核糖的含量在4.9~368.6 mg/g之间。此外,在这些样品中检测到了果糖,之前未见文献报道。不同样品中糖含量的差异可能与螺旋藻的生长环境有关,还需进一步的研究。

表 2 12种糖的线性范围、回归方程、相关系数(r)、检出限和定量限
Y: peak area;X: mass concentration, mg/L.

表 3 螺旋藻样品中12种糖的加标回收率及精密度(n=5)

表 4 实际螺旋藻样品的检测结果(n=3)
ND: not detected.
3 结论
本研究建立了一种快速、准确检测12种糖类化合物的超高效液相色谱-串联质谱分析方法。该方法样品前处理简单,准确度高。应用于螺旋藻样品的检测结果表明,多种单糖在不同样品中含量差异较大。该方法的建立可为阐明螺旋藻多糖的结构组成及其活性提供技术支撑及基础数据。
[1] Cai B N, Chen C X, Chen X, et al. Food Science, 2013, 34(23): 83
蔡冰娜, 陈纯馨, 陈忻, 等. 食品科学, 2013, 34(23): 83
[2] Chen T, Wong Y S, Zheng W. Phytochemistry, 2006, 67(22): 2424
[3] Chen X E, Fang X B, Yu H, et al. Journal of Zhejiang Ocean University, 2008, 27(4): 361
陈小娥, 方旭波, 余辉, 等. 浙江海洋学院学报, 2008, 27(4): 361
[4] Yang Y J, Jiang R Z, Chen Y H, et al. Chinese Traditional Patent Medicine, 2005, 27(6): 706
杨勇杰, 姜瑞芝, 陈英红, 等. 中成药, 2005, 27(6): 706
[5] Liu X H, Chen Y G, Lin L, et al. Food Science and Technology, 2009, 34(9): 270
刘晓涵, 陈永刚, 林励, 等. 食品科技, 2009, 34(9): 270
[6] Zhang J, Li C Y, Li J P, et al. Central South Pharmacy, 2012, 10(6): 421
张杰, 李春艳, 李劲平, 等. 中南药学, 2012, 10(6): 421
[7] Chen J, Li J, Sun A D, et al. Ind Crop Prod, 2014, 60: 138
[8] Zakharova A M, Grinshtein I L, Kartsova L A. J Anal Chem, 2013, 68(12): 1081
[9] Wan Q, Wu X H, Fan H J, et al. Journal of Instrumental Analysis, 2014, 33(11): 1231
万强, 吴学昊, 范华均, 等. 分析测试学报, 2014, 33(11): 1231
[10] Ren H N, Chen X H, Bi K S, et al. Journal of Shenyang Pharmaceutical University, 2009, 26(3): 206
任浩娜, 陈晓辉, 毕开顺, 等. 沈阳药科大学学报, 2009, 26(3): 206
[11] Shanmugavelan P, Su Y K, Kim J B, et al. Carbohyd Res, 2013, 380: 112
[12] Ma C, Sun Z, Chen C, et al. Food Chem, 2014, 145(10): 784
[13] Käkölä J M, Alén R J, Isoaho J P, et al. J Chromatogr A, 2008, 1190: 150
[14] Ding Y, Yu H, Mou S. J Chromatogr A, 2002, 982(2): 237
[15] Bignaridi C, Cavazza A, Corradini C. Int J Carbohyd Chem, 2012, 2012: 1155
[16] Wang C P, Zhu L, Liu X, et al. Food Science, 2014, 35(20): 164
王川丕, 诸力, 刘新, 等. 食品科学, 2014, 35(20): 164
[17] Ben Y G, Zhong H M, Li K, et al. Chinese Traditional Patent Medicine, 2011, 33(6): 1078
贲永光, 钟红茂, 李康, 等. 中成药, 2011, 33(6): 1078
[18] Zhao P, Li W H, Zhu Z H, et al. Food Science, 2009, 30(20): 151
赵鹏, 李稳宏, 朱骤海, 等. 食品科学, 2009, 30(20): 151
Determination of monosaccharide inSpirulinapolysaccharide by ultra performance liquid chromatography-tandem mass spectrometry
ZHAO Dan1,2, FENG Feng2, SU Youzhi3, ZHANG Jingnan1,YU Lian1, SU Jin1*, ZHANG Feng2*
(1.CollegeofPharmacy,JiamusiUniversity,Jiamusi154000,China;2.InstituteofFoodSafety,ChineseAcademyofInspection&Quarantine,Beijing100176,China;3.YiliEntry-ExitInspectionandQuarantineBureau,Yining835000,China)
An ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method was developed for the determination of 12 carbohydrates in hydrolysate ofSpirulinaincluding rhamnose, xylose, arabinose, fructose, mannose, glucose, galactose, mannitol, ribose, fucose, glucuronic acid and galacturonic acid. Samples were extracted with deionized water using ultrasonic-assisted extraction (UAE) and hydrolysis by trifluoroacetic acid (TFA). The hydrolysate was separated on a Waters Acquity BEH Aminde column (100 mm×2.1 mm, 1.7 μm) using 10 mmol/L ammonium formate/water-10 mmol/L ammonium formate/acetonitrile solution as the mobile phase. The electrospray ionization tandem quadrupole mass spectrometric analysis was carried out in the negative ion mode using multiple reaction monitoring (MRM). The limits of detection were 0.005-0.15 mg/kg and the linear ranges were 0.05-5 mg/L. The average recoveries were 80.21%-121.6% at the spiked levels of the carbohydrates contents of 50%, 100% and 150% found in samples. The method was applied to analyze the samples ofSpirulina. Fucose, galactose, arabinose, rhamnose, glucose, fructose, xylose, and ribose were detected in most of the samples, and their contents were in the range of 0.3-889.4 mg/g. The type and content of carbohydrates that can be detected in the sample were significantly different. Common characteristic peaks were identified as fucose, galactose, arabinose, rhamnose, glucose, fructose, xylose, ribosethe. However, mannitol and mannose were not detected in all samples. This developed method could provide technical support and basic data for the study of structure and activity of polysaccharide fromSpirulina.
ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS); monosaccharide; polysaccharide;Spirulina; quality evaluation
10.3724/SP.J.1123.2016.09038
2017-09-20
国家自然科学基金项目(81274101).
Foundation item: National Natural Science Foundation of China (No. 81274101).
O658
A
1000-8713(2017)04-0413-08
* 通讯联系人. E-mail:sj0129@163.com(苏瑾);E-mail:fengzhang@126.com(张峰).
