黄东海-西北太平洋大气颗粒态有机胺浓度及粒径分布特征分析❋
2017-04-10于沛然郭天锋朱玉姣高会旺姚小红
于沛然, 郭天锋, 朱玉姣, 高会旺, 姚小红
(中国海洋大学海洋环境与生态教育部重点实验室,山东 青岛 266100)
黄东海-西北太平洋大气颗粒态有机胺浓度及粒径分布特征分析❋
于沛然, 郭天锋, 朱玉姣, 高会旺, 姚小红
(中国海洋大学海洋环境与生态教育部重点实验室,山东 青岛 266100)
在2014和2015年春季分别搭载黄东海-西北太平洋航次采集分粒径气溶胶样品,从浓度水平、空间分布及粒径分布三方面,对海洋大气气溶胶中颗粒态有机胺进行分析。DMA+(二甲胺)和TMA+(三甲胺)为采样区域内主要存在的颗粒态有机胺。两种颗粒态有机胺在不同海域和年份有较大的浓度变化。空间分布结果表明,海洋大气颗粒态有机胺主要来源于海洋而非陆地传输,且与海洋生物活动性存在密切关系。大部分样品中颗粒态有机胺主要分布在0.1~1.8 μm,主要来源于气粒凝结、非均相反应、一次燃烧排放以及云过程几种形成过程,各形成过程在不同样品中相对重要性各不相同。在2015年航次中还观测到0.01~0.056 μm存在明显模态分布,且在一些低浓度样品中这一模态甚至和积聚模态对颗粒态有机胺的贡献相当。
海洋大气气溶胶;有机胺;粒径分布;黄东海;西北太平洋
海洋大气气溶胶作为地球生态系统中重要的一环,它可以通过吸收或散射太阳辐射对全球气候系统产生直接影响,也可以通过形成云凝结核和冰核对气候产生间接影响[1]。有机胺作为海洋大气有机组成中的一种,对海洋二次有机气溶胶(SOA)的贡献仅次于二甲基硫(DMS),远高于其他挥发性有机物[2]。同时,由于自身较强的碱性和良好的水溶性,有机胺还可以通过酸碱反应对海洋大气气溶胶酸度产生影响[3-4]。此外,理论研究和实验室实验均表明有机胺还可以在新粒子的形成和颗粒物的增长中起到重要作用[5-8]。
目前,对于有机胺的研究多为理论和实验室研究,而海洋作为有机胺的主要自然源之一,有关海洋大气环境中有机胺的观测研究还相对较少,尤其是存在潜在气候效应的颗粒态有机胺。Sorooshian等在东太平洋的观测研究发现,海洋大气颗粒物中的二乙胺(DEA)和甲基磺酸(MSA)浓度的升高和海水中高叶绿素浓度相一致。此外,还指出了气溶胶成分和粒径分布对云凝结核活性的重要作用,但并未对海洋大气颗粒态有机胺粒径分布进行具体研究[9]。Muller等在北大西洋的观测研究指出,在海洋大气细颗粒中高浓度有机胺主要存在于0.14~0.42 μm[10]。Hu等对中国近海大气气溶胶研究也指出海洋大气颗粒态有机胺浓度与叶绿素浓度存在良好的相关关系,且对0.43~11 μm颗粒态有机胺粒径分布情况进行分析[11]。在已有的研究中由于采样器切割粒径范围较粗且缺少小于0.1 μm超细粒径段颗粒态有机胺的分布情况,在一定程度上限制了我们对海洋大气气溶胶中有机胺的认识,这两点限制将在本研究中得到较好的解决。
黄东海是全球最为典型的陆架浅海之一,初级生产力相对较高,可能存在丰富的有机胺来源,同时,西北太平洋海域是代表性的寡营养开阔大洋海域。从黄东海到西北太平洋是研究海洋大气颗粒态有机胺浓度水平、理化特性及形成机制的理想区域。本研究在2014—2015年搭载两次春季黄东海-西北太平洋科学考察航次,使用nano-MOUDI分级采样器收集分粒径大气气溶胶样品,并采用离子色谱法对颗粒物中的常规离子及有机胺进行定量分析。通过对气溶胶样品中颗粒态有机胺的浓度水平和粒径分布研究,意在探究黄东海-西北太平洋大气颗粒态有机胺的主要来源及形成过程。
1 研究方法
1.1 采样方法
海洋大气气溶胶样品采样点设置在东方红2号科考船顶楼气象室,采样口距离海面约10 m。在2014年春季航次中,使用11级nano-MOUDI采样器,切割粒径为:18、10、5.6、3.2、1.8、1.0、0.56、0.32、0.18、0.10和0.056 μm。使用预先高温灼烧过(600℃,6 h)的石英膜作为采样膜。在2015年春季航次中,使用14级nano-MOUDI采样器,切割粒径分别为:18、10、5.6、3.2、1.8、1.0、0.56、0.32、0.18、0.10、0.056、0.032、0.018和0.010 μm。1~11级使用聚四氟乙烯采样滤膜(47 mm, PALL Life Sciences),12~14级使用有ZefluorTM支撑的聚四氟乙烯滤膜(90 mm, PALL Life Sciences)。2个航次中采样流量均为29.4 L·min-1。在停船和顺风行驶(视风向90°到270°)时,为避免样品受船舶排放的影响,仪器关闭,同样下雨时仪器也会被关闭。2014年航次中,采样膜一天更换一次,由于停船的影响,实际采样时间从几小时到十几小时不等。在2015年航次中采样膜1~3 d更换一次。每个航次中均有2~3套空白样采集。采集好的气溶胶样品用事先高温灼烧过的铝箔包裹(450 ℃,6 h),放入样品盒中,置于-20℃冰箱避光保存,待分析。
1.2 分析方法
在化学分析前,首先需对样品进行预处理,使用0.5 mL无水乙醇和20 mL去离子水(18.2 MΩ·cm),在冰水混合条件下,对样品进行超声萃取20min。随后,将萃取液通过0.45 μm滤头(PTFE,25 mm, PALL Life Sciences)过滤,去除难溶性杂质,滤出液置于离心管中待测。

2 结果与讨论
2.1 浓度水平
为对比中国近海和开阔大洋大气颗粒态有机胺的浓度水平,2014和2015年2个航次均被划分为2个航段,黄东海航段和西北太平洋航段。分析结果显示,DMA+(二甲胺)和TMA+(三甲胺)为黄东海-西北太平洋大气气溶胶中主要存在的颗粒态有机胺。在2014年春季航次中,由于所采用石英膜本身的性质,海洋大气中气态有机胺可能被直接吸附到石英膜上,致使测得浓度偏高,造成采样正误差。如表1所示,在黄东海PM10大气颗粒物中DMA+和TMA+测得浓度分别为(1.3±0.68)和(0.56±0.50)nmol·m-3,在西北太平洋航段2种有机胺的浓度分别为(0.63±0.41)和(1.3±1.1) nmol·m-3,测得浓度为颗粒态有机胺及直接吸附到石英膜上气态有机胺浓度之和。2015年采样使用聚四氟乙烯采样膜,采样不受气态有机胺的影响。在黄东海PM10大气颗粒物中DMA+和TMA+的浓度分别为(0.40±0.36)和(0.38±0.42) nmol·m-3,在西北太平洋海域,DMA+和TMA+两种有机胺浓度分别为(0.28±0.16)和(0.22±0.12) nmol·m-3。在西北太平洋海域PM1大气颗粒物中DMA+的浓度与已发表在阿拉伯海[12]、北大西洋[13]以及东地中海[14]测得DMA+的浓度可比,但TMA+并未获得有效的可比数据。
对比2014和2015年2个航次2种有机胺浓度可以发现,不管在黄东海或者西北太平洋海域,2014年样品中有机胺的浓度均高于2015年。DMA+的浓度在黄东海海域是2015年黄东海的约3倍,在西北太平洋海域浓度约为2015年的2倍。对于TMA+,2014年样品在黄东海和西北太平洋海域的浓度分别是2015年样品的1.6和6倍。为探究2014和2015 2个航次较大的浓度差异是否来源于气态有机胺的影响,本文对2014年航次中气态有机胺浓度进行估算。大气环境中气态的有机胺不受切割粒径的限制,对各个级的影响一致。所以,在2014年采样中,作者以每套样品中浓度最低的一级作为气态有机胺的浓度,即气溶胶采样正误差。由此可得,在2014年黄东海海域大气环境中气态DMA和TMA的浓度分别为,(0.05±0.05)和(0.01±0.03) nmol·m-3。在西北太平洋海域DMA和TMA的浓度分别为(0.03±0.03)和(0.03±0.06) nmol·m-3。气态有机胺对各级采样器均有影响,所以在PM10粒径段每级采样器均扣除气态有机胺影响后,本文得到接近真实的PM10颗粒态有机胺浓度,在黄东海DMA+和TMA+浓度分别为(0.83±0.37)和(0.43±0.27) nmol·m-3,在西北太平洋海域2种颗粒态有机胺的浓度分别为,(0.32±0.19)和(1.03±0.72) nmol·m-3。由此可见,在2014年春季航次中采样区域内气态有机胺的浓度远小于颗粒态。此外,对比2个航次,2014年样品中颗粒态有机胺均高于2015年样品,尤其在2014年西北太平洋海域TMA+浓度为2015年的3倍。
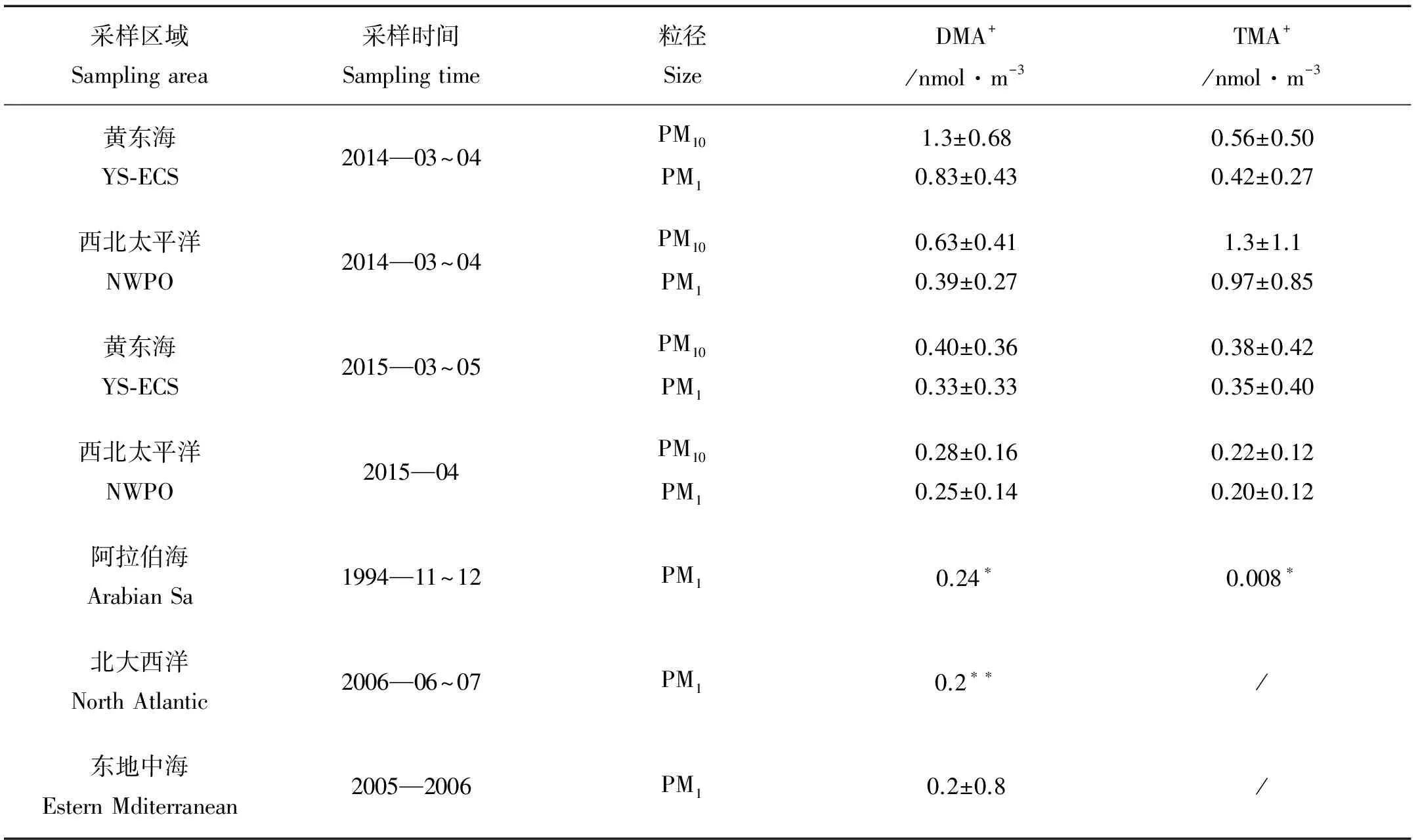
表1 本研究及已发表研究中DMA+和TMA+浓度汇总
注: “*”表示均值; “**”表示中值;其它表示均值±标准偏差; “/”表示浓度未给出。
“*” means average value, “**” means median value, others mean average±STDEV value, “/” means not given.

2.2 空间分布

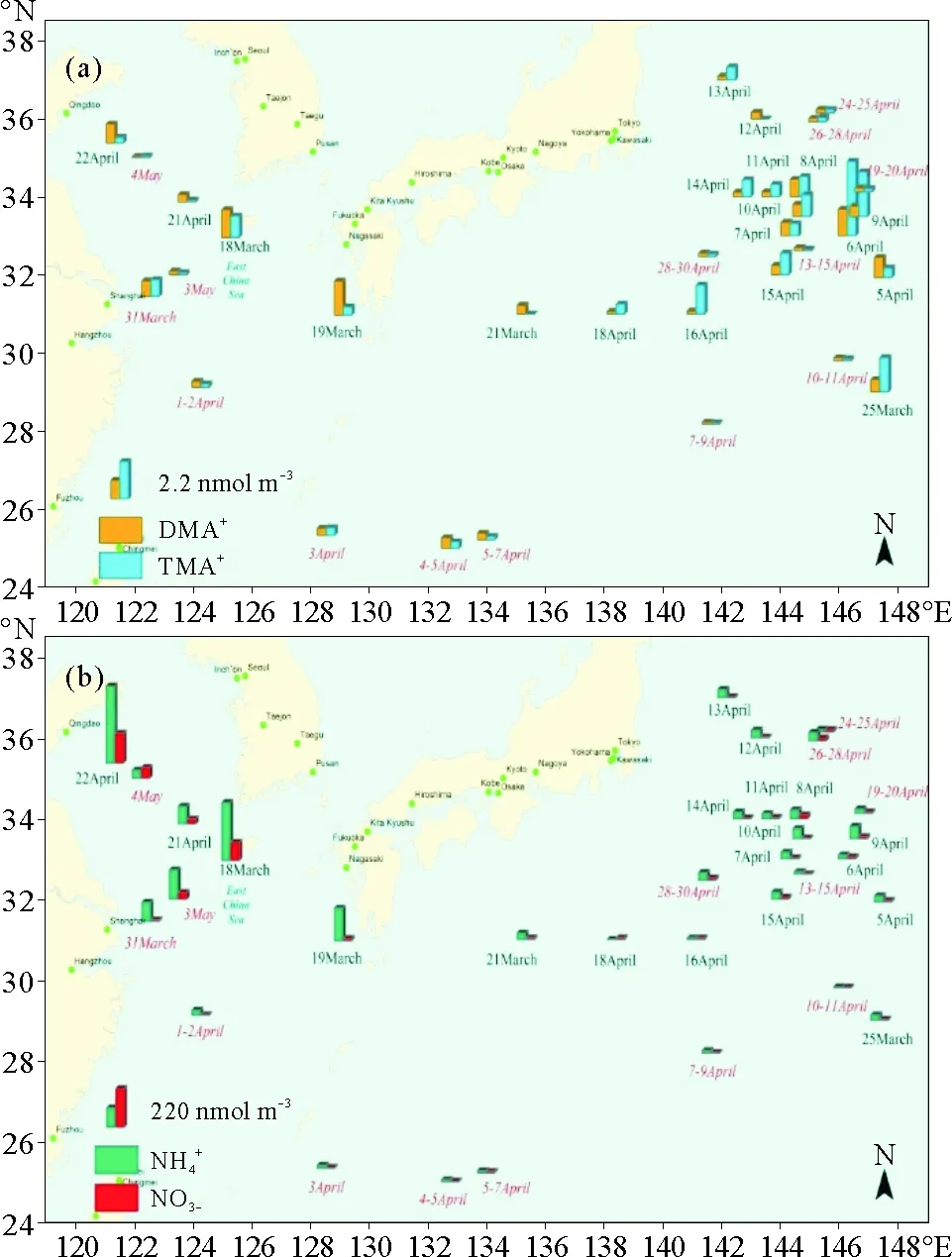
(a:DMA+和TMA+;和;红色斜体样品编号为2015年样品品。a: DMA+and TMA+; b:; The samples of 2015 cruise campaign were marked in red and italic.c.)
已有研究表明,海水中有机胺的释放与海洋生物活动性存在密切的关系[13, 19-20]。在本研究中选择海水叶绿素a的浓度,来探究其和大气中颗粒态有机胺的关系。在2014年春季航次中,结果表明,2种颗粒态有机胺的浓度与叶绿素浓度卫星数据有较好的对应关系(见图2)。在3月25日到4月16日采集到较高浓度有机胺样品,同时在相对应的采样区域内3月30日到4月14日叶绿素浓度对比3月23日到29日有明显的升高。在文献[11]中也发现了DMA+和TMA+两种有机胺和叶绿素荧光值存在良好的相关性。

(a:3月23—29日;b:3月30—4月6日;c:4月7—14日。a: 23~29 March; b:30 March~6 April; c: 7~14 April.)
为得到更准确的叶绿素浓度数据,进一步研究采样区域内两种有机胺和叶绿素的关系,在2015年春季航次中作者获得了叶绿素实测数据,具体空间分布如图3所示。在整个航次中,叶绿素浓度也呈现较大的空间变化,最为明显的是,由于丰富营养物质的输入,位于长江入海口海域的3月31日样品中叶绿素浓度最高,相对应的气溶胶样品中DMA+和TMA+浓度也为该航次中的最高值。但在其他样品中,叶绿素浓度并不能与有机胺浓度有很好的对应关系。例如,在4月1—2日的样品中,叶绿素浓度与3月31日接近,但DMA+浓度则小1.5倍,TMA+浓度小3.5倍。对整个航次中叶绿素浓度与DMA+和TMA+浓度进行相关性分析结果表明,叶绿素浓度与DMA+和TMA+浓度的相关系数分别为R2=0.40和R2=0.41。 这可能是由于在不同生长期海洋微生物对碳源、氮源等营养物质吸收不同,新陈代谢活动也不相同有关,所以大气中颗粒态有机胺浓度与叶绿素浓度水平未能取得良好相关关系。Bradley等研究表明在海水中浮游植物爆发的衰亡期会有大量的氮释放到大气环境中[21]。因此,在浮游植物衰亡期可能存在大量有机胺的释放,而在衰亡期采得的样品可能才会获得颗粒态有机胺与叶绿素的良好相关关系。虽然海洋大气中有机胺的释放与海洋生物活动性存在密切的关系,但与叶绿素浓度并没有直接的关系。
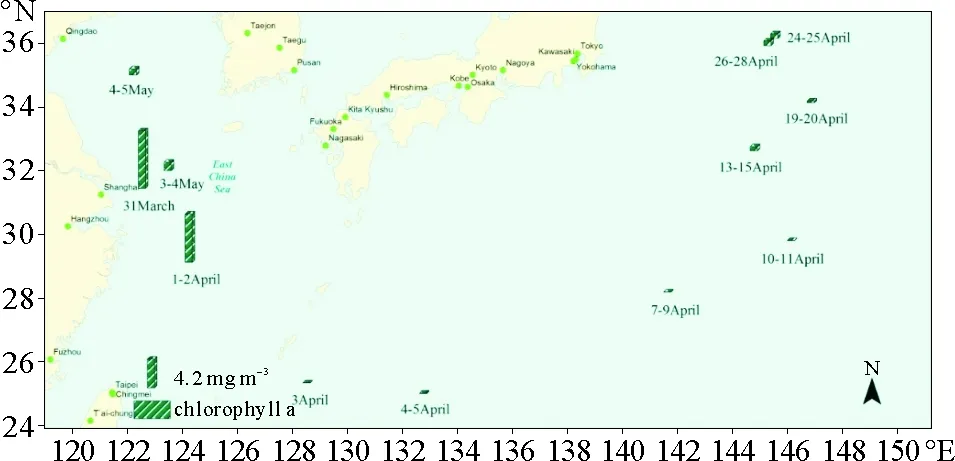
图3 2015年春季航次叶绿素浓度空间分布Fig.3 Geographical distributions of Chlorophyll-a concentration in spring cruise campaign in 2015
2.3 粒径分布
不同形成过程产生的颗粒物分布在不同的粒径段上,因此,可以通过对颗粒态有机胺粒径分布分析推断颗粒态有机胺的形成过程以及各种形成过程对颗粒态有机胺的贡献。在2014年春季航次黄东海航段中,DMA+和TMA+均主要分布在0.1~1.8 μm,存在一个占主导地位的积聚模态,但两者峰值所处的位置有所不同。DMA+峰值位于0.32~0.56 μm,而TMA+的峰值则出现在0.18~0.32 μm。不同的粒径分布代表两种颗粒态有机胺可能来源于不同的形成过程,已有研究表明,在0.4 μm左右的颗粒物来自于非均相反应或者一次燃烧排放,而0.2 μm左右的颗粒物来源于气粒凝结作用[22]。在西北太平洋航段中,DMA+呈现出与黄东海航段相似的粒径分布,主要分布在0.1~1.8 μm,存在一个占主导地位的积聚模态,略有不同的是,此航段中DMA+在0.18~0.32 μm和0.32~0.56 μm两级采样器上均有较高的浓度,表明非均相反应或一次燃烧排放和气粒凝结作用对DMA+贡献相当。与DMA+和黄东海航段的两种颗粒态有机胺均有所不同,TMA+呈现出随切割粒径不断减小浓度逐渐上升的粒径分布规律。西北太平洋海域较高浓度的TMA+可能与这种粒径分布形式存在一定关系,具体原因还需要更多的观测研究论证。


(a,b:2014年黄东海;c,d:2014年西北太平洋;e,f:2015年黄东海;g,h:2015年西北太平洋。a,b: DMA+ and TMA+ over YS-ECS in 2014; c,d: DMA+ and TMA+ over NWPO in 2014; e,f: DMA+ and TMA+ over YS-ECS in 2015; g,h: DMA+ and TMA+ over NWPO in 2015.)
在2015年春季航次中,2种颗粒态有机胺在2个海域大气中粒径分布较为一致。DMA+和TMA+均主要分布在0.1~1.8 μm,存在一个峰值在0.32~0.56 μm的占主导地位的积聚模态。表明在此航次中,2种有机胺均主要来源于非均相反应或一次燃烧排放。与2014年有所不同的是,在2015年航次中使用14级采样器,获得了颗粒态有机胺在10~56 nm的有效数据。此外,此航次中使用聚四氟乙烯采样膜,样品不会受到气态有机胺的影响。因此,我们使用对数正态分布函数对样品粒径分布模态进行逐个拟合[23],结果如图5所示。
结果表明,DMA+和TMA+在大部分样品中均存在一个占主导地位的(0.4±0.1)μm的积聚模态,主要来源于非均相反应或一次燃烧排放。此外,DMA+在4月19—20日、5月3—4日和5月4—5日拟合结果呈现出在0.1~1 μm两个相当的模态。同样,TMA+在4月19—20日、4月28—30日和5月4—5日也存在这样的模态分布。DMA+和TMA+在(0.2±0.1)和(0.7±0.1)μm两个相当的模态分别属于凝结模态和液滴模态,分别来自于气粒凝结作用和云过程[22, 24-25]。此外,大部分样品在小于0.1 μm还存在一个贡献较小但清晰可见的模态,且在一些低浓度样品中,这一模态对颗粒态有机胺的贡献与积聚模态相当,甚至可以在粒径分布中占主导地位。这一结果表明,有机胺可能在纳米粒径段酸性物质的中和以及颗粒物的形成和增长中起到重要作用。


图5 2015年黄东海-西北太平洋大气颗粒物中DMA+和TMA+对数正态分布函数模态拟合
3 结论
(1)DMA+和TMA+是黄东海和西北太平洋海域主要存在的两种颗粒态有机胺,且浓度水平在不同海域和不同年份均有较大的变化。2014年DMA+和TMA+在黄东海和西北太平洋海域浓度均高于2015年航次,虽然由于采样膜的原因,2014年样品存在采样正误差,但两种有机胺在气态中浓度较低,影响相对较小。
(2)黄东海和西北太平洋海域大气颗粒态有机胺主要来源于海洋而非长距离陆地传输。海洋大气环境中的有机胺与海洋生物活动性存在密切的关系,但与叶绿素浓度并没有直接关系,在浮游植物爆发的衰亡期更有可能存在大量有机胺的释放。
(3)采样区域内颗粒态有机胺主要分布在(0.1~1.8)μm,存在(0.2±0.1)、(0.4±0.1)及(0.7±0.1)μm几个主要模态,分别来自于气粒凝结作用、非均相反应或一次燃烧排放和云过程,在不同样品中几种形成过程的贡献有所不同。
(4)DMA+和TMA+在10~100 nm存在明显的模态,且在一些低浓度样品中,这一纳米粒径段模态对颗粒态有机胺的贡献与积聚模态相当甚至高于积聚模态,说明有机胺在纳米粒径段酸性物质的中和及新颗粒的形成中起到重要作用。
[1] Ge X, Wexler A S, Clegg S L. Atmospheric amines-Part I. A review[J]. Atmospheric Environment, 2011, 45(3): 524-546.
[2] Myriokefalitakis S, Vignati E, Tsigaridis K, et al. Global modeling of the oceanic source of organic aerosols[J]. Advances in Meteorology, 2010,939171,doi.10.1155/2010/939171.
[3] Williams B J, Goldstein A H, Kreisberg N M, et al. Major components of atmospheric organic aerosol in southern California as determined by hourly measurements of source marker compounds[J]. Atmospheric Chemistry and Physics, 2010, 10(23): 11577-11603.
[4] Lee Y N, Weber R, Ma Y, et al. Airborne measurement of inorganic ionic components of fine aerosol particles using the particle‐into‐liquid sampler coupled to ion chromatography technique during ACE‐Asia and TRACE‐P[J]. Journal of Geophysical Research: Atmospheres, 2003, 108(D23): 2932-2938.
[5] Kirkby J, Curtius J, Almeida J, et al. Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation[J]. Nature, 2011, 476(7361): 429-433.
[6] Kulmala M, Kontkanen J, Junninen H, et al. Direct observations of atmospheric aerosol nucleation[J]. Science, 2013, 339(6122): 943-946. [7] Berndt T, Stratmann F, Sipil? M, et al. Laboratory study on new particle formation from the reaction OH+ SO 2: influence of experimental conditions, H 2 O vapour, NH 3 and the amine tert-butylamine on the overall process[J]. Atmospheric Chemistry and Physics, 2010, 10(15): 7101-7116.
[8] Wang L, Khalizov A F, Zheng J, et al. Atmospheric nanoparticles formed from heterogeneous reactions of organics[J]. Nature Geoscience, 2010, 3(4): 238-242.
[9] Sorooshian A, Padró L T, Nenes A, et al. On the link between ocean biota emissions, aerosol, and maritime clouds: Airborne, ground, and satellite measurements off the coast of California[J]. Global Biogeochemical Cycles, 2009, 23(4):GB4007,doj:10.1029/2009 GB003464.
[10] Müller C, Iinuma Y, Karstensen J, et al. Seasonal variation of aliphatic amines in marine sub-micrometer particles at the Cape Verde islands[J]. Atmospheric Chemistry and Physics, 2009, 9(24): 9587-9597.
[11] Hu Q, Yu P, Zhu Y, et al. Concentration, Size Distribution, and Formation of Trimethylaminium and Dimethylaminium Ions in Atmospheric Particles over Marginal Seas of China*[J]. Journal of the Atmospheric Sciences, 2015, 72(9): 3487-3498.
[12] Gibb S W, Mantoura R F C, Liss P S. Ocean‐atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea[J]. Global Biogeochemical Cycles, 1999, 13(1): 161-178.
[13] Facchini M C, Decesari S, Rinaldi M, et al. Important source of marine secondary organic aerosol from biogenic amines[J]. Environmental Science & Technology, 2008, 42(24): 9116-9121.
[14] Violaki K, Mihalopoulos N. Water-soluble organic nitrogen (WSON) in size-segregated atmospheric particles over the Eastern Mediterranean[J]. Atmospheric Environment, 2010, 44(35): 4339-4345.
[15] Silva P J, Erupe M E, Price D, et al. Trimethylamine as precursor to secondary organic aerosol formation via nitrate radical reaction in the atmosphere[J]. Environmental Science & Technology, 2008, 42(13): 4689-4696.
[16] Savoie D L, Prospero J M, Merrill J T, et al. Nitrate in the atmospheric boundary layer of the tropical South Pacific: Implications regarding sources and transport[J]. Journal of Atmospheric Chemistry, 1989, 8(4): 391-415.
[17] Savoie D L, Prospero J M, Nees R T. Nitrate, non-sea-salt sulfate, and mineral aerosol over the northwestern Indian Ocean[J]. Journal of Geophysical Research: Atmospheres, 1987, 92(D1): 933-942.
[18] Schlesinger W H, Hartley A E. A global budget for atmospheric NH3[J]. Biogeochemistry, 1992, 15(3): 191-211.
[19] Calderón S M, Poor N D, Campbell S W. Estimation of the particle and gas scavenging contributions to wet deposition of organic nitrogen[J]. Atmospheric Environment, 2007, 41(20): 4281-4290.[20] Wang X, Lee C. Sources and distribution of aliphatic amines in salt marsh sediment[J]. Organic Geochemistry, 1994, 22(6): 1005-1021.
[21] Bradley P B, Sanderson M P, Nejstgaard J C, et al. Nitrogen uptake by phytoplankton and bacteria during an induced Phaeocystis pouchetii bloom, measured using size fractionation and flow cytometric sorting[J]. Aquatic Microbial Ecology, 2010, 61(1): 89-104.[22] Seinfeld J H, Pandis S N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change[M]. America: John Wiley & Sons, 2012.[23] Yu H, Yu J Z. Polycyclic aromatic hydrocarbons in urban atmosphere of Guangzhou, China: Size distribution characteristics and size-resolved gas-particle partitioning[J]. Atmospheric Environment, 2012, 54: 194-200.
[24] Rehbein P J, Jeong C, Mcguire M L, et al. Cloud and fog processing enhanced gas-to-particle partitioning of trimethylamine[J]. Environmental Science & Technology, 2011, 45(10): 4346-4352.[25] Yao X H, Zhang L. Supermicron modes of ammonium ions related to fog in rural atmosphere[J]. Atmospheric Chemistry and Physics, 2012, 12(22): 11165-11178.
责任编辑 庞 旻
Concentration and Size Distribution of Amines in Marine Atmospheric Particles over Yellow Sea, East China Sea and Northwest Pacific Ocean
YU Pei-Ran, GUO Tian-Feng, ZHU Yu-Jiao, GAO Hui-Wang, YAO Xiao-Hong
(The Key Lab of Marine Environmental Science and Ecology,Ministry of Education, Ocean University of China, Qingdao 266100, China)
In this study, concentration, spatial distribution and size distribution of particulate amines in marine aerosol were investigated during two cruises from the Yellow Sea (SYS) and East China Sea (ECS) to the Northwest Pacific Ocean (NWPO). DMA+(dimethylaminium) and TMA+(trimethylaminium) were the main detected aminium in the sampling area.The concentrations of DMA+and TMA+varied in different years and sampling area.The spatial distribution result indicated that the particulate amines in the marine atmosphere were originated from marine source other long range transport from the continent. The emission of amines was possibly related to biogenic activities in the seas. Size distribution data showed that DMA+and TMA+in most of samples dominantly existed in the size range of 0.18~1.8 μm during these campaigns. The fitted modal patterns implied that they could be due to gas-particle condensation, heterogeneous reactions or primary combustion and cloud processing of aerosols.Relative importance of these formation pathways varied from samples to samples. In addition, a clear mode at 0.01~0.056 μm was observed in some samples in 2015, and this mode was even comparable to the accumulation mode in 0.1~1.8 μm in some low concentration samples.
Marine aerosol; amines; size distribution; Yellow Sea and East China Sea; Northwest Pacific Ocean
国家重点基础研究发展计划项目(2014CB953700)资助 Supported by National Program on Key Basic Research Project(2014CB953700)
2016-04-21;
2016-05-19
于沛然(1990-),男,硕士生。Email:noelyu@foxmail.com
X142
A
1672-5174(2017)05-019-08
10.16441/j.cnki.hdxb.20160141
于沛然,郭天锋,朱玉姣,等.黄东海-西北太平洋大气颗粒态有机胺浓度及粒径分布特征分析[J].中国海洋大学学报(自然科学版), 2017, 47(5): 19-26.
YU Pei-Ran,GUO Tian-Feng,ZHU Yu-Jiao,et al.Concentration and size distribution of amines in marine atmospheric particles over Yellow Sea, East China Sea and Northwest Pacific Ocean[J].Periodical of Ocean University of China, 2017, 47(5): 19-26.
