Tissue-type plasminogen activator is a homeostatic regulator of synaptic function in the central nervous system
2017-04-07,
,
1 Department of Neurology & Center for Neurodegenerative Disease, Emory University School of Medicine, Atlanta, GA, USA
2 Department of Neurology, Veterans Affairs Medical Center, Atlanta, GA, USA
3 Division of Neurosciences, Yerkes National Primate Research Center, Atlanta, GA, USA
Tissue-type plasminogen activator is a homeostatic regulator of synaptic function in the central nervous system
Valerie Jeanneret1, Manuel Yepes1,2,3,*
1 Department of Neurology & Center for Neurodegenerative Disease, Emory University School of Medicine, Atlanta, GA, USA
2 Department of Neurology, Veterans Affairs Medical Center, Atlanta, GA, USA
3 Division of Neurosciences, Yerkes National Primate Research Center, Atlanta, GA, USA
How to cite this article:Jeanneret V, Yepes M (2017) Tissue-type plasminogen activator is a homeostatic regulator of synaptic function in the central nervous system. Neural Regen Res 12(3):362-365.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Funding:This work has been supported in part by National Institutes of Health Grants NS-079331 (to MY) and NS-091201 (to MY).
Membrane depolarization induces the release of the serine proteinase tissue-type plasminogen activator (tPA) from the presynaptic terminal of cerebral cortical neurons. Once in the synaptic cleft this tPA promotes the exocytosis and subsequent endocytic retrieval of glutamate-containing synaptic vesicles, and regulates the postsynaptic response to the presynaptic release of glutamate. Indeed, tPA has a bidirectional effect on the composition of the postsynaptic density (PSD) that does not require plasmin generation or the presynaptic release of glutamate, but varies according to the baseline level of neuronal activity. Hence, in inactive neurons tPA induces phosphorylation and accumulation in the PSD of the Ca2+/calmodulin-dependent protein kinase IIα (pCaMKIIα), followed by pCaMKIIα-induced phosphorylation and synaptic recruitment of GluR1-containing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. In contrast, in active neurons with increased levels of pCaMKIIα in the PSD tPA induces pCaMKIIα and pGluR1 dephosphorylation and their subsequent removal from the PSD. These effects require active synaptic N-methyl-D-aspartate (NMDA) receptors and cyclin-dependent kinase 5 (Cdk5)-induced phosphorylation of the protein phosphatase 1 (PP1) at T320. These data indicate that tPA is a homeostatic regulator of the postsynaptic response of cerebral cortical neurons to the presynaptic release of glutamate via bidirectional regulation of the pCaMKIIα /PP1 switch in the PSD.
tissue-type plasminogen activator (tPA); homeostatic plasticity; Ca2+/calmodulin-dependent protein kinase; post-synaptic density; protein phosphatase 1; plasmin
Introduction
Tissue-type plasminogen activator (tPA) is a serine proteinase that in the intravascular space has a fibrinolytic role mediated by its ability to catalyze the conversion of plasminogen into plasmin (Camiolo et al., 1971; Collen and Lijnen, 1991). Earlier studies found that tPA is abundantly expressed in the brain (Sappino et al., 1993), and although it was initially believed that endothelial cells were the only source of this tPA, later work indicated that tPA is also found in glial cells (Siao et al., 2003) and neurons (Nicole et al., 2001; Yepes et al., 2009). Since then tPA has been implicated in a plethora of functions in the brain including the development of synaptic plasticity (Qian et al., 1993; Madani et al., 1999; Oray et al., 2004; Yepes et al., 2016), the detection and adaptation to metabolic stress (Wu et al., 2012), modulation of blood-brain barrier (BBB) permeability (Yepes et al., 2003), and remodeling of the extracellular matrix (ECM) (Berardi et al., 2004).
A further advancement in the understanding of tPA’s function in the brain was attained by the observation that its release from the presynaptic terminal of glutamatergic neurons (Gualandris et al., 1996) triggers the synaptic vesicle cycle in cerebral cortical neurons (Wu et al., 2015). More specifically, it was discovered that membrane depolarization induces the rapid secretion of tPA at extrasynaptic sites, and that by promoting the recruitment of the cytoskeletal protein βII-spectrin to the active zone (AZ), tPA enlarges the synaptic release site. At the same time, it was found that tPA also induces the phosphorylation of synapsin I, a protein that clusters glutamate-containing synaptic vesicles (SVs) in the reserve pool of the presynaptic terminal. This effect is of pivotal importance for neurotransmision, because phosphorylation of synapsin I at serine 9 allows SVs to translocate from the reserve pool of synaptic vesicles to the synaptic release site, already enlarged by tPA, where they release their load of excitatory neurotransmitters into the synaptic cleft (Wu et al., 2015). Remarkably, this effect does not lead to depletion of SVsfrom the presynaptic terminal, because tPA also promotes their endocytic retrieval from the presynaptic membrane,viacalcineurin-mediated dynamin I dephosphorylation and the formation of the actin scaffold necessary for the newly formed SVs to re-enter the synaptic vesicle cycle (Wu et al., 2015).
These observations originated two important questions about the synaptic effect of the release of tPA from the presynaptic terminal of cerebral cortical neurons: first, if tPA promotes the exocytosis and endocytic retrieval of glutamate-containing SVs, then is tPA an inductor of excitotoxicity by perpetuating the synaptic release of glutamate? Second, does tPA released from the axonal bouton have an effect on the postsynaptic terminal?
Effect of tPA on the Composition of the Post-Synaptic Density
To address these questions, we used electron microscopy to study the effect of tPA on the postsynaptic density (PSD), an electron-dense structure attached to the postsynaptic terminal that undergoes rapid changes in molecular composition, structure, and function in response to variations in synaptic activity (Dosemeci et al., 2001). We found that either treatment with recombinant tPA (rtPA) or the release of neuronal tPA is followed by a rapid increase in the thickness of the PSD (Jeanneret et al., 2016). Ten we used phosphoproteomics and biochemical studies with extracts of the PSD to identify the mechanism underlying this effect. Surprisingly, we found that treatment with rtPAin vitroor the endogenous release of neuronal tPA prompted by a brief episode of cerebral ischemiain vivo, causes a rapid and sustained increase in the expression of Ca2+/Calmodulin-dependent protein kinase IIα phosphorylated at T286 (pCaMKII) in the PSD.
CaMKIIα is a serine-threonine kinase that is highly abundant in the PSD (Petersen et al., 2003). The influx of calcium into the post-synaptic terminal of the active synapse leads to calcium/calmodulin-mediated CaMKIIα activation that is followed by its translocation from the postsynaptic actin cytoskeleton to the PSD where it binds to N-methyl-D-aspartate receptors (NMDARs). Intriguingly, upon its phosphorylation at T286, CaMKIIα remains active even after calcium concentrations fall to baseline levels (Miller and Kennedy, 1986; Lisman et al., 2012). This “autonomy” has led many to postulate CaMKIIα as a “memory molecule” with a pivotal role in learning and the development of synaptic plasticity (Lisman et al., 2012). Importantly, it was shown that tPA-induced CaMKIIα phosphorylation and accumulation in the PSD is independent of its effect on the presynaptic release of glutamate (Jeanneret et al., 2016).
Despite the relevance of these events for the development of synaptic plasticity under physiological conditions, it is also important to note that several groups have reported that cerebral ischemia causes pCaMKIIα phosporylation, and that its accumulation in the PSD leads to neuronal death (Coultrap et al., 2011; Liu et al., 2012; Lu et al., 2013). Tus, as stated above, it is conceivable to propose that by inducing its accumulation in the PSD, tPA has a neurotoxic effect. To study this possibility, we used a model in which either exogenous (rtPA), or endogenous (neuronal tPA), was added to neurons in which pCaMKIIα was already accumulated in the PSD. Surprisingly, we found that tPA does not cause a further increase in pCaMKIIα expression in the PSD of neurons with high baseline levels of the kinase, but that instead it decreases them to those found under baseline, non stimulated conditions (Jeanneret et al., 2016). In summary, these findings indicate that tPA has a bidirectional effect on the phosphorylation and accumulation of pCaMKIIα in the PSD that depends on the baseline levels of the kinase. Tus, while in previously inactive neurons tPA increases pCaMKIIα expression in the PSD, in those that are already active tPA has the opposite effect, decreasing its accumulation in the postsynaptic terminal. Importantly, although the effect of tPA on pCaMKIIα is mediated by synaptic NMDARs, it does not require the conversion of plasminogen into plasmin (Jeanneret et al., 2016).
tPA Regulates the CamKIIalpha/PP1 Switch in the Postsynaptic Density
CaMKII phosphorylation is tightly regulated by a family of serine/threonine protein phosphatases (PP) (Colbran, 2004). There are different PPs, with PP1, 2A, 2B and 2C accounting for the most of the phosphatase activity in the brain. PP1 is the PP of the PSD, and our data indicate that tPA induces its phosphorylation at T320viaCdk5 activation. Together, these results show that tPA has a bidirectional effect on the PP1/CaMKIIα switch.
tPA is a Homeostatic Regulator of Synaptic Activity
To understand the physiological importance of this effect we used a proteomics approach to study the effect of tPA-induced CaMKIIα phosphorylation on synaptic function. We found that tPA induces CaMKIIα-mediated phosphorylation at S831 and its subsequent recruitment to the PSD of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) subunit GluR1. Again, as previously noted, this is a bidirectional effect in which tPA induces GluR1 phosphorylation (pGluR1) in previously inactive neurons and pGluR1 dephosphorylation in neurons with high baseline levels of synaptic activity (Jeanneret et al., 2016). The physiological importance ofthese findings is underscored by the observation that glutamatergic synapses that contain NMDA but not AMPA receptors are “silent”. However, they become“active” following pCaMKIIα-induced phosphorylation and synaptic recruitment of AMPA receptors contining GluR1 subunits (Liao et al., 2001). Together, these data indicate that while tPA decreases the activity of those synapses that are already active, it increases it in those that are previously inactive. These results confirm our initial observation that tPA is a homeostatic regulator of synaptic function.
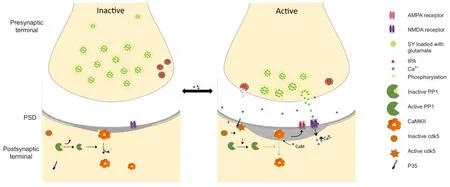
Figure 1 Tissue-type plasminogen activator (tPA) induces homeostatic plasticity in cerebral cortical neurons.
Homeostatic plasticity is the process whereby neural circuits maintain their activity levels within a constant range (Turrigiano and Nelson, 2000; Turrigiano, 2008). This is a mechanism that allows networks to develop compensatory mechanisms aimed at maintaining the homeostatic balance of a population of synapses (Turrigiano, 2008). For instance, to maintain the equilibrium of the system, a prolonged decrease in neuronal activity activates compensatory mechanisms that increase the population of AMPA receptors and spine size on the postsynaptic terminal. The results discussed here indicate that tPA is an effective inductor of homeostatic plasticity in cerebrocortical neurons during physiological and pathological conditions.
In summary, the data currently available allow to propose a model in which tPA is an inductor of homeostatic plasticityviaits ability to bidirectionally regulate the PP1/ CaMKIIα switch (Figure 1). This is of particular importance for our understanding of the mechanisms underlying the development of synaptic plasticity and for the potential therapeutic use of rtPA to protect the synapse of patients with neurological diseases that such as cerebral ischemia are characterized by the presence of severe synaptic dysfunction.
Author contributions:VJ wrote the manuscript. MY conceived the idea and wrote the manuscript.
Conflicts of interest:None declared.
Berardi N, Pizzorusso T, Maffei L (2004) Extracellular matrix and visual cortical plasticity: freeing the synapse. Neuron 44:905-908.
Camiolo SM, Thorsen S, Astrup T (1971) Fibrinogenolysis and fibrinolysis with tissue plasminogen activator, urokinase, streptokinase-activated human globulin, and plasmin. Proc Soc Exp Biol Med 138:277-280.
Colbran RJ (2004) Protein phosphatases and calcium/calmodulin-dependent protein kinase II-dependent synaptic plasticity. J Neurosci 24:8404-8409.
Collen D, Lijnen HR (1991) Basic and clinical aspects of fibrinolysis and thrombolysis. Blood 78:3114-3124.
Coultrap SJ, Vest RS, Ashpole NM, Hudmon A, Bayer KU (2011) CaMKII in cerebral ischemia. Acta Pharmacol Sin 32:861-872.
Dosemeci A, Tao-Cheng JH, Vinade L, Winters CA, Pozzo-Miller L, Reese TS (2001) Glutamate-induced transient modification of the postsynaptic density. Proc Natl Acad Sci U S A 98:10428-10432.
Gualandris A, Jones TE, Strickland S, Tsirka SE (1996) Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurosci 16:2220-2225.
Jeanneret V, Wu F, Merino P, Torre E, Diaz A, Cheng L, Yepes M (2016) Tissue-type plasminogen activator (tPA) modulates the postsynaptic response of cerebral cortical neurons to the presynaptic release of glutamate. Front Mol Neurosci 9:121.
Liao D, Scannevin RH, Huganir R (2001) Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci 21:6008-6017.
Lisman J, Yasuda R, Raghavachari S (2012) Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci 13:169-182.
Liu Z, Xu J, Shen X, Lv C, Xu T, Pei D (2012) CaMKII antisense oligodeoxynucleotides protect against ischemia-induced neuronal death in the rat hippocampus. J Neurol Sci 314:104-110.
Lu Q, Harris VA, Sun X, Hou Y, Black SM (2013) Ca(2)(+)/calmodulin-dependent protein kinase II contributes to hypoxic ischemic cell death in neonatal hippocampal slice cultures. PLoS One 8:e70750.
Madani R, Hulo S, Toni N, Madani H, Steimer T, Muller D, Vassalli JD (1999) Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J 18:3007-3012.
Miller SG, Kennedy MB (1986) Regulation of brain type II Ca2+/ calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell 44:861-870.
Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A (2001) The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med 7:59-64.
Oray S, Majewska A, Sur M (2004) Dendritic spine dynamics are regulated by monocular deprivation and extracellular matrix degradation. Neuron 44:1021-1030.
Petersen JD, Chen X, Vinade L, Dosemeci A, Lisman JE, Reese TS (2003) Distribution of postsynaptic density (PSD)-95 and Ca2+/ calmodulin-dependent protein kinase II at the PSD. J Neurosci 23:11270-11278.
Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D (1993) Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature 361:453-457.
Sappino AP, Madani R, Huarte J, Belin D, Kiss JZ, Wohlwend A, Vassalli JD (1993) Extracellular proteolysis in the adult murine brain. J Clin Invest 92:679-685.
Siao CJ, Fernandez SR, Tsirka SE (2003) Cell type-specific roles for tissue plasminogen activator released by neurons or microglia after excitotoxic injury. J Neurosci 23:3234-3242.
Turrigiano GG (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135:422-435.
Turrigiano GG, Nelson SB (2000) Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol 10:358-364.
Wu F, Torre E, Cuellar-Giraldo D, Cheng L, Yi H, Bichler EK, Garcia PS, Yepes M (2015) Tissue-type plasminogen activator triggers the synaptic vesicle cycle in cerebral cortical neurons. J Cereb Blood Flow Metab 35:1966-1976.
Wu F, Wu J, Nicholson AD, Echeverry R, Haile WB, Catano M, An J, Lee AK, Duong D, Dammer EB, Seyfried NT, Tong FC, Votaw JR, Medcalf RL, Yepes M (2012) Tissue-type plasminogen activator regulates the neuronal uptake of glucose in the ischemic brain. J Neurosci 32:9848-9858.
Yepes M, Roussel BD, Ali C, Vivien D (2009) Tissue-type plasminogen activator in the ischemic brain: more than a thrombolytic. Trends Neurosci 32:48-55.
Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA (2003) Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest 112:1533-1540.
Yepes M, Wu F, Torre E, Cuellar-Giraldo D, Jia D, Cheng L (2016) Tissue-type plasminogen activator induces synaptic vesicle endocytosis in cerebral cortical neurons. Neuroscience 319:69-78.
*Correspondence to: Manuel Yepes, M.D., myepes@emory.edu.
10.4103/1673-5374.202924
Accepted: 2017-02-28
杂志排行

中国神经再生研究(英文版)的其它文章
- Telomerase and mTOR in the brain: the mitochondria connection
- The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson’s disease
- Neuromelanin, one of the most overlooked molecules in modern medicine, is not a spectator
- Novel insights into the role of NF-κB p50 in astrocytemediated fate specification of adult neural progenitor cells
- Anesthetic considerations for patients with acute cervical spinal cord injury
- Impacts of the retinal environment and photoreceptor type on functional regeneration