基于计算机辅助水解的中药大豆寡肽的ETA拮抗活性预测
2017-03-28乔连生蒋芦荻雒刚刚路芳陈艳昆王
乔连生+蒋芦荻+雒刚刚+路芳+陈艳昆+王灵芝+李贡宇+张燕玲
[摘要] 中药寡肽是中药发挥药效的关键组分之一,系统地研究中药寡肽的组成及其药效是中药物质基础及作用机制研究的关键。该研究拟基于计算机辅助水解和分子对接等模拟技术,以中药大豆为研究载体,解析其降压寡肽成分,并预测其潜在的内皮素受体A(endothelin receptor A,ETA)拮抗活性。该文通过收集大豆中的储存蛋白序列,基于计算机辅助水解方法进行蛋白的模拟消化水解,并将水解获得的寡肽构建虚拟结构数据库。随后该研究构建了ETA肽类拮抗剂的药效团模型,包括1个疏水特征、1个负电中心、1个芳环特征和5个排除体积。同时,利用同源模建方法构建ETA的蛋白三维结构模型,并将其用于分子对接研究,采用一致性打分评价化合物的ETA拮抗活性。通过构建ETA肽类拮抗剂的药效团模型、同源模建的三维蛋白结构以及分子对接模型,共预测获得27条可能具有潜在ETA拮抗活性的寡肽分子。该文进一步分析关键氨基酸GLN165,并结合文献,说明其对拮抗剂活性的重要性。计算机辅助水解方法可以高效地对已知序列结构的中药蛋白进行模拟水解,结合分子模拟模型,能进一步辨识中药寡肽潜在的生物学活性。该研究为快速、高效开展中药来源的肽类物质的活性机制研究提供了方法学依据。
[关键词] 计算机辅助水解; 寡肽; 大豆; 內皮素受体A; 药效团; 同源模建; 分子对接
[Abstract] Oligopeptides are one of the the key pharmaceutical effective constituents of traditional Chinese medicine(TCM). Systematic study on composition and efficacy of TCM oligopeptides is essential for the analysis of material basis and mechanism of TCM. In this study, the potential anti-hypertensive oligopeptides from Glycine max and their endothelin receptor A (ETA) antagonistic activity were discovered and predicted based on in silico technologies.Main protein sequences of G. max were collected and oligopeptides were obtained using in silico gastrointestinal tract proteolysis. Then, the pharmacophore of ETA antagonistic peptides was constructed and included one hydrophobic feature, one ionizable negative feature, one ring aromatic feature and five excluded volumes. Meanwhile, three-dimensional structure of ETA was developed by homology modeling methods for further docking studies. According to docking analysis and consensus score, the key amino acid of GLN165 was identified for ETA antagonistic activity. And 27 oligopeptides from G. max were predicted as the potential ETA antagonists by pharmacophore and docking studies.In silico proteolysis could be used to analyze the protein sequences from TCM. According to combination of in silico proteolysis and molecular simulation, the biological activities of oligopeptides could be predicted rapidly based on the known TCM protein sequence. It might provide the methodology basis for rapidly and efficiently implementing the mechanism analysis of TCM oligopeptides.
[Key words] in silico proteolysis; oligopeptides; Glycine max; ETA; pharmacophore; homology modeling; docking
中药蛋白是中药中一类常见的活性物质,具有明显而特殊的药理活性[1]。从两千多年前阿胶的应用,到水蛭素的制备和大豆多肽的上市,中药蛋白和生物活性肽的应用具有悠久的历史,已有大量的药性、药理和临床数据。而中药蛋白具有物理化学性质不稳定、易水解和易氧化的特点,因此,基于传统的中药化学方法,通过对中药蛋白提取分离酶解获取中药活性肽,往往具有分离困难、耗时费力的特点。而计算机辅助水解是基于生物信息学方法,预测蛋白质序列在特定蛋白酶或化学物质在给定条件下的切割位点的技术,该技术具有快速高效可靠的寡肽预测能力[2],已较为广泛的应用于食品工业研究[3-4]。
中药大豆是豆科植物大豆Glycine max的成熟种子,现代研究表明大豆活性肽具有良好的降压活性[5]。内皮素受体A(endothelin receptor A,ETA)是治疗高血压和肺动脉高压的经典靶标,其第一代拮抗剂即为肽类化合物[6]。因此,基于大豆寡肽开发ETA拮抗剂具有较高的研究价值。本研究拟利用计算机辅助水解技术,进行中药大豆的模拟消化水解,并结合基于药效团和分子对接的虚拟筛选技术,预测中药大豆寡肽的ETA拮抗活性。
1 材料与方法
1.1 大豆蛋白的模拟水解 本研究在Uniport(http://www.uniprot.org/)中检索已解析的大豆种子储存蛋白的多肽序列,共获得5条大豆球蛋白(glycinin)和3条伴大豆球蛋白(conglycinin)序列,主要包括大豆球蛋白G,大豆球蛋白G1-G4,β-伴大豆球蛋白的α,α′和β链。其为大豆中的主要储存蛋白,占分离蛋白的80%以上[7]。基于获得的蛋白序列,本研究采用BIOPEP(http://www.uwm.edu.pl/biochemia/index.php/pl/biopep)进行计算机辅助模拟水解,以预测蛋白序列的切割位点。本研究采用模拟胃肠道的水解策略,选择胃蛋白酶(pH 1.3)、胰蛋白酶和糜蛋白酶作为消化酶[8]。针对获得的寡肽序列,利用Discovery Studio 4.0(DS)进行寡肽三维结构数据库的构建。
1.2 ETA肽类拮抗剂的药效团模型的构建 为了更可靠的筛选ETA的肽类拮抗剂,本研究拟采用DS中的3D-QSAR pharmacophore(Hypogen)模块,构建ETA肽类拮抗剂的药效团模型。构建药效团的训练集和测试集中的50个活性化合物均来源于文献报道[9],其包括直链肽和环肽2类化学结构,其活性值分布5个数量级。
对训练集中20个化合物在CHARMm中进行三维构象生成和能量最小化处理。随后,对其进行药效特征分析,发现活性化合物中包括的药效特征有氢键受体(hydrogen bond acceptor,A)、亲脂性氢键受体(hydrogen bond acceptor lipid,Ali)、氢键供体(hydrogen bond donor,D)、疏水基团(hydrophobic region,H)、脂性疏水基团(hydrophobic aliphatic region,Hal)、芳香疏水基团(hydrophobic aromatic region,Har)、芳香环(ring aromatic,R)、负电基团(ionizable negative group,N)。
基于特征分析,进一步构建药效团模型。首先,对训练集中活性化合物进行构象分析,采用Best模式,设置能量收敛值为20 kcal·mol-1,最大构象数目255个。选择生成的5个药效特征为氢键受体、氢键供体、疏水基团、芳香环和负电基团,同时,模型中通过增加5个排除体积(maximum excluded volumes,Ev)以提高模型的特异性,并设置化合物的活性不确定度为1.5,利用Hypogen模块构建10个候选药效团模型。
针对生成的药效团模型需进行进一步的模型评价。利用剩余的30个活性化合物及从Binding Database(http://www.bindingdb.org/bind/index.jsp)中随机挑选的90个非活性化合物,以1∶3的比例生成测试集,并對测试集分子进行同训练集分子相同的计算处理。随后,利用生成的候选药效团模型与测试集化合物进行匹配筛选,计算相应的有效命中率(hit rate of active compounds,HRA)、辨识有效性指数(identify effective index,IEI)、综合评价指数(comprehensive appraisal index,CAI)等指标对药效团优劣进行评价[10]。3个外部评价指标计算公式如下所示(1)~(3),其中TD代表测试集中化合物总数,TA代表测试集中活性化合物数目。Ha代表命中的活性化合物数目,Ht代表命中化合物总数。本文综合利用药效团模型的多种评价参数,选出最优的药效团模型。利用最优的药效团模型进行大豆寡肽库的虚拟筛选,其中搜索模式为Best Search、匹配模式为柔性匹配,初步获得潜在的活性寡肽。
HRA=Ha/TA×100%(1)
IEI=(Ha/Ht)/(TA/TD)(2)
CAI=IEI×HRA(3)
1.3 ETA的同源模建及分子对接 ETA(Uniprot/P25101)由427个氨基酸残基组成,是典型的G蛋白偶联受体(G protein-coupled receptor,GPCR)中A家族的7次跨膜蛋白。通过对已解析的同家族蛋白晶体进行序列比对,分析其同源性和相似性的高低,选择相似性较高的蛋白作为模板蛋白。本研究同源模建使用Modeller 9.13程序,采用多模板建模方法,生成5个候选模型进行进一步优化。首先,基于逐级能量最小化方法,先约束所有重原子,优化氢原子,再约束保守跨膜区,优化侧链及Loop区,最后优化跨膜区。能量最小化过程采用最陡下降法和共轭梯度法进行优化。最后基于拉氏图和ERRAT分析,利用SAVES服务器(http://services.mbi.ucla.edu/SAVES/)对同源模建蛋白进行评价。
结合对蛋白空腔的自动识别,同时参考GPCR同家族活性口袋的位置,定义ETA蛋白的活性口袋。由于ETA活性口袋中可能存在多种结合模式[11],为保证分子对接结果的准确性,本研究选择较大的活性口袋半径。本研究利用DS中的Libdock模块进行分子对接。首先,以阳性药作为测试,分别对接进入活性口袋中,并同时计算5种打分函数,采用一致性打分的方法,评价对接模型的可靠性,并分析ETA肽类拮抗剂可能存在的作用模式。随后,利用上述的对接模型进一步筛选药效团的命中结果,筛选潜在的ETA肽类拮抗剂。
2 结果与讨论
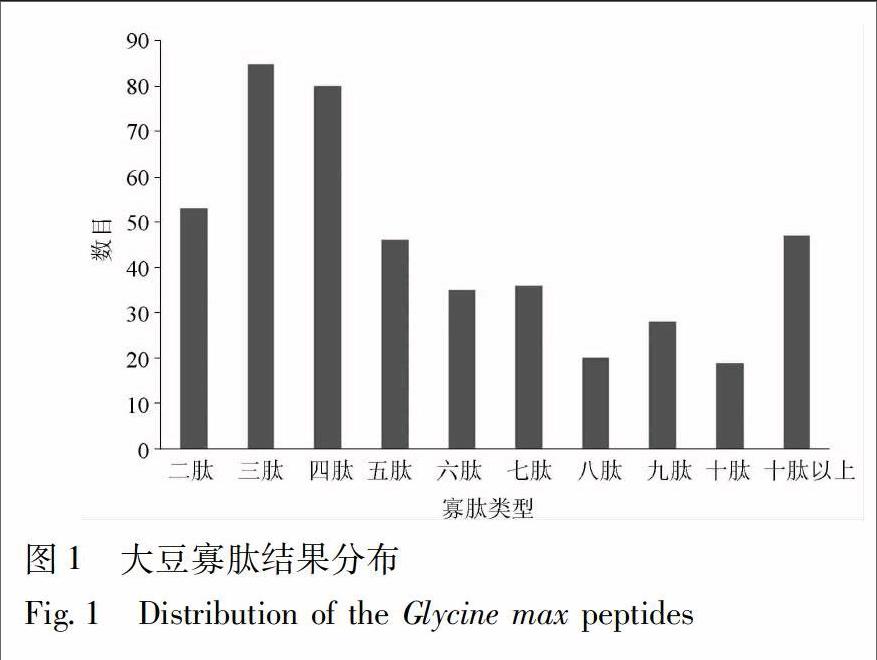
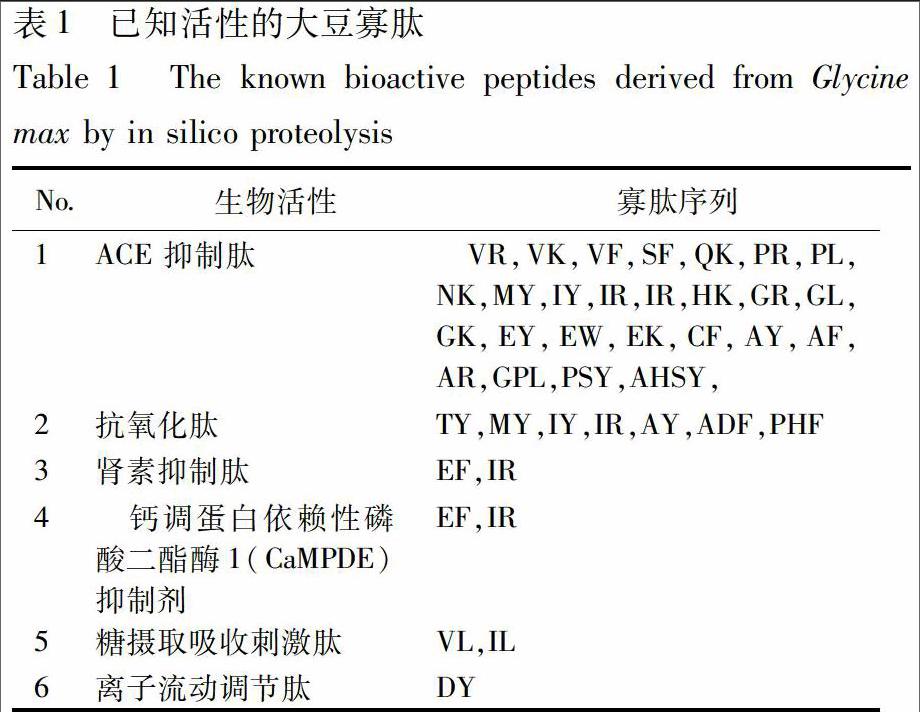
2.1 大豆寡肽库的构建 通过模拟水解,本研究共获得449条多肽,其中各段寡肽的数目分布见图1,由于2~6肽可能具有较好的相对分子质量与成药性[2],所以选择2~6肽共299条作为本研究的主要研究对象。通过对模拟水解结果与已知的活性肽数据库BIOPEP比对,共发现已知活性肽32条,其主要的生物学活性包括ACE抑制作用和抗氧化作用等,其结果见表1,表明大豆寡肽具有较为广泛的药理活性。
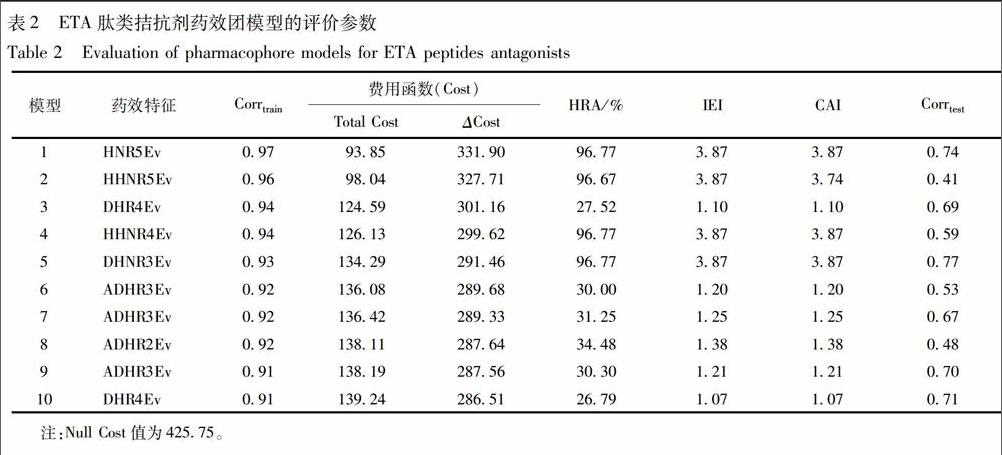
2.2 ETA肽类拮抗剂药效团模型的建立与评价 基于Hypogen的药效团建模方法主要包括2类内部评价参数,即训练集活性化合物的预测结果与真实活性的相关系数Corrtrain,以及费用函数(Cost)对所构建的药效团模型的显著性评价。本研究构建的10个药效团内部评价参数见表2。模型的Null Cost为425.75,ΔCost为Null Cost与Total Cost的差值,ΔCost大于60说明模型有90%以上的显著性。
利用测试集对10个候选药效团进行评价,计算相应外部评价参数,包括药效团的活性化合物命中率HRA、辨识有效性指数IEI和综合评价指数CAI以及测试集中活性化合物的预测值与真实值之间的相关系数Corrtest,其结果见表2。本研究生成的药效团模型主要包括2类,即HNR型和DHR型。HNR型主要包括1个疏水特征,1个负电中心和1个芳环基团,而DHR型主要包括1个氢键受体,1个疏水特征和1个芳香基团。通过对比其CAI值差异,发现HNR型具有更好的测试集筛选能力。进一步分析药效团对测试集的活性预测能力,发现1号和5号药效团具有相当的活性化合物辨识能力。通过比较2个药效团的空间结构差异,发现2个药效团中HNR 3个特征之间的空间距离及角度具有相似性,同时,5号药效团的D特征与其排除体积相重叠,因此最终选择1号药效团作为最优药效团对大豆寡肽库进行虚拟筛选,结果共获得64条寡肽。其中,VVF与药效团具有较好的匹配,其匹配值为0.85,见图2。
2.3 ETA同源模建及分子对接研究 通过同源比对,选择3个蛋白晶体结构作为模板进行同源模建,包括Delta Opioid Receptor(PDB:4N6H_A),Neurotensin Receptor 1(PDB:4BV0_A)和orphanin FQ receptor(PDB:4EA3_A)。3个受体均来源于GPCR的A家族,且其与ETA的序列相似度 (similarity)分别为40.0%,39.8%,38.8%,其一致性(identity)分别为26%,24%,25%。利用获得的3个模板进行多模板建模,通过能量最小化处理,利用拉氏图和ERRAT评价模型的优劣,最终模型和评价参数见图3。其中,拉氏图的最适残基量为96.96%,ERRAT的值为91.41,显示模建结构具有良好的立体化学参数和空间结构。
ETA分子对接结果:本研究通过分析ETA的空腔结构,自动识别了8个活性口袋,通过文献中关键氨基酸的描述和参考GPCR同家族的位点情况,最终确定对接口袋,其半径为15 。先将训练集中20个阳性寡肽与活性口袋对接,20个化合物均能对接进入活性口袋中,进一步计算Jain,-PLP2,-PMF,-PMF04和-PLP1 5种打分函数,综合已有的LibDockScore打分,进行一致性打分评价[12]。其中,一致性打分大于3的化合物为95.0%,说明了对接模型和一致性打分的可靠性[13]。随后,分析ETA与阳性药发生相互作用的高频氨基酸,产生氢键作用的氨基酸主要包括:LYS329,ARG326,ASP351和GLN165,產生疏水作用的氨基酸主要有LEU347,LYS140,LEU141,ARG145,ARG326,LYS329,与文献报道基本一致[14-15]。
进一步将测试集中30个活性分子与活性口袋对接,一致性打分大于3的化合物为96.7%,说明对接模型和一致性打分评价对化合物具有较好的预测能力。发生氢键作用的关键氨基酸主要有GLN165,LYS329和ASP351,产生疏水作用的高频氨基酸有ARG326,LEU322和LYS329等,与文献报道及训练集化合物基本一致。其中高活性化合物Com1385与ETA之间的相互作用见图4。
利用构建的分子对接模型和一致性打分方法,筛选被药效团命中的64条寡肽。27条寡肽的一致性打分为6,提示大豆虚拟水解获得的寡肽可能具有较好的ETA拮抗活性。其中,VVF能与关键氨基酸具有良好的匹配,VVF中苯丙氨酸的羧基与药效团的负电中心匹配,而其羰基氧与GLN165发生氢键相互作用,而其羟基氢能与ASP351发生氢键相互作用,因此,VVF的苯丙氨酸羧基可能是发挥活性的重要基团[16],其结果见图5。
3 结论
本研究基于计算机辅助水解方法,对大豆中的主要蛋白进行模拟消化水解,获得2~6肽的寡肽基本结构共299条。在此基础上,通过构建ETA肽类拮抗剂的药效团,以及ETA蛋白同源模建与分子对接,筛选并预测大豆寡肽的ETA拮抗活性,共获得27条可能具有ETA拮抗活性的寡肽分子,其结果有待进一步的活性验证。本文通过基于生物信息学和计算机辅助药物设计的方法,从大豆蛋白中筛选潜在的ETA拮抗活性寡肽,为探讨中药蛋白发挥药效的作用机制探讨提供了一条新的研究方法,为开展中药蛋白的酶解分离或药理验证提供指导。
[参考文献]
[1] 邹吉利,徐南平.中药活性多肽研究进展[J].湖北中医药大学学报, 2012, 14(4):66.
[2] 邹平. 基于生物信息学与QSAR及分子对接的菜粕活性肽筛选及活性研究[D]. 杭州:浙江大学, 2014.
[3] Dziuba B, Dziuba M. New milk protein-derived peptides with potential antimicrobial activity: an approach based on bioinformatic studies[J]. Int J Mol Scis, 2014, 15(8):14531.
[4] 孙国威,乐国伟,施用晖.模拟酶解大豆7S、11S蛋白及其抗氧化活性的研究[J].食品工业科技, 2010(7):101.
[5] 于婷婷,韩飞,陈光.大豆降压肽研究进展[J].粮油食品科技, 2008, 16(2):27.
[6] 杨菁.内皮素受体拮抗剂的设计与合成[D].北京:中国人民解放军军事医学科学院,2008.
[7] 田琨,管娟,邵正中,等.大豆分离蛋白结构与性能[J].化学进展, 2008, 20(4):565.
[8] Lafarga T, O′Connor P, Hayes M. Identification of novel dipeptidyl peptidase-IV and angiotensin-I-converting enzyme inhibitory peptides from meat proteins using in silico analysis[J]. Peptides, 2014, 59(9):53.
[9] Fukami T, Yamakawa T, Niiyama K, et al. Synthesis and structure-activity relationships of 2-substituted D-tryptophan-containing peptidic endothelin receptor antagonists: importance of the C-2 substituent of the D-tryptophan residue for endothelin A and B receptor subtype selectivity[J]. J Med Chem, 1996, 39(12):2313.
[10] Jiang L, Zhang X, Chen X, et al. Virtual screening and molecular dynamics study of potential negative allosteric modulators of mGluR1 from Chinese herbs[J]. Molecules, 2015, 20:12769.
[11] Mey J G R D, Compeer M G, Pieter L, et al. ETA -receptor antagonists or allosteric modulators[J]. Trends Pharmacol Sci, 2011, 32(6):345.
[12] 劉维国.抗药物依赖功效药效团模型的构建及其虚拟筛选[D].北京:中央民族大学, 2013.
[13] Krovat E M, Langer T. Impact of scoring functions on enrichment in docking-based virtual screening: an application study on renin inhibitors[J]. J Chem Inf Comp Sci, 2004, 44(3): 1123.
[14] Breu V, Hashido K, Broger C, et al. Separable binding sites for the natural agonist endothelin-1 and the non-peptide antagonist bosentan on human endothelin-A receptors[J]. Eur J Biochem, 1995, 231(1):266.
[15] Jens L, Alexander O, Michael B, et al. Structural determinants for selective recognition of peptide ligands for endothelin receptor subtypes ETA and ETB[J]. J Pept Sci, 2009, 15(7):479.
[16] Khuraijam Dhanachandra S, Karthikeyan M. Molecular modeling, quantum polarized ligand docking and structure-based 3D-QSAR analysis of the imidazole series as dual AT1 and ETA receptor antagonists[J]. Acta Pharm Sin, 2013, 34(12):1592.
