昆虫基因组及数据库研究进展
2017-03-16尹传林李美珍丁思敏郭殿豪
尹传林,李美珍,贺 康,丁思敏,郭殿豪,席 羽,李 飞
(浙江大学昆虫科学研究所,杭州310058)
昆虫基因组及数据库研究进展
尹传林,李美珍,贺 康,丁思敏,郭殿豪,席 羽,李 飞*
(浙江大学昆虫科学研究所,杭州310058)
基因组序列为昆虫分子生物学研究提供丰富的数据资源,推动系统生物学在古老的昆虫学中蓬勃发展。昆虫基因组学研究已经成为当前的研究热点,目前在NCBI登录注册的昆虫基因组测序计划有494项,其中已提交原始测序数据的昆虫有225种,完成基因组拼接的有215种,具有基因注释的有65种,公开发表的昆虫基因组有43篇。本文综述了测序技术发展的历史及其对昆虫基因组研究的推动作用、昆虫基因组的组装和注释及其存在的问题、昆虫基因组测序进展、昆虫基因组数据库的发展及基因数据挖掘利用的基本思路和对策,以及昆虫基因大数据在害虫防治和资源昆虫利用中的应用前景。
昆虫基因组;组装与注释;数据挖掘与分析;基因组数据库;害虫防治;资源昆虫利用
昆虫是生物界种类数量最多、最古老的类群之一,距今3.5亿年的古生代泥盆纪就已出现,大约构成所有生物种类的50%左右 (Robinson,etal., 2011),目前已经被描述鉴定的昆虫种类有一百万多种。作为重要的活化石,昆虫的进化研究可以探秘生命的起源以及地球环境的变更。昆虫与人类的活动息息相关,既有令人烦恼的农业害虫和卫生害虫,也有让人赏心悦目的观赏昆虫。农业生态系统离不开昆虫,地球上75%以上的开花植物都依靠昆虫来授粉 (Robinsonetal., 2011)。昆虫学作为一门独立的分支进入科学领域,迄今已有300多年历史。
随着测序技术的快速发展,在生物大数据的潮流下,古老的昆虫学逐渐迈入基因组时代。昆虫学者利用各种组学研究手段如基因组、转录组、蛋白组、代谢组等产生了大量的生物数据,从系统生物学的角度来解决昆虫学研究中的问题,为昆虫学研究带来了新的视角,焕发了新的生机。本文围绕昆虫基因组学研究中的组装、注释、数据挖掘和基因数据库等方面进行了综述,对目前存在的问题进行了总结,对未来的发展趋势进行了展望。
1 测序技术的进步和生物信息学的发展
昆虫基因组学研究得益于测序技术的巨大进步和生物信息学的逐渐普及。测序技术根据其发展的历史可以分为三个不同的时代:以链终止法或链降解法为原理的一代测序技术(如Sanger测序技术)、以边合成(边链接)边测序为原理的二代测序技术(主要包括ABI公司的SOLiD技术、Illumina公司的Solexa技术和Roche公司的454技术等),以及单分子测序的三代测序技术(如PacBio公司的SMRT技术和Oxford Nanopore公司的纳米孔单分子测序技术等)(Heatheretal., 2016)(图1)。
1975年由桑格(Sanger)和考尔森(Coulson)发明的链终止法 (Sangeretal., 1975),以及1976年由马克西姆(Maxam)和吉尔伯特(Gilbert)发明的链降解法 (Maxametal., 1977),开启了核酸测序的新纪元。利用第一代测序技术,测定了噬菌体X174的基因组序列,全长5375个碱基,这是首个生命体的基因组序列 (Sangeretal., 1977)。2001年,利用Sanger测序技术完成了人类基因组计划 (Venteretal., 2001)。果蝇是第一个被测序的昆虫 (Adamsetal., 2000),之所以被优先选择进行基因组测序,是因为果蝇一直被视为生命科学研究中最重要的模式生物之一。但其实更重要的原因,是果蝇基因组比较小(仅180 Mb左右),可以用来检测全基因组鸟枪法(Whole Geome Shotgun, WGS)在人类基因组测序中的可行性。在没有其他测序技术可供选择情况下,第一代Sanger测序技术是唯一的技术主角,其具有明显的优势,读长最高可达1000 bp,准确性高达99.999%。然而,其缺点也十分明显,测序成本过高,通量低,无法实现真正的大规模应用。
在科研需求和市场利润的双重驱动下,催生了3个重要的二代测序技术(SOLiD技术、Solexa技术和454技术)。在人类基因组测序计划要惊动各国领导人的时代,美国NIH启动了“1000美元基因组计划”,资助2亿美金来推动测序技术的进步。正是这种前瞻性的资助计划,改写了生命科学研究的进程,也是当前生命科学各个研究领域的基因组计划发展如火如荼的重要基础。第二代测序技术极大地降低了测序成本,提高了测序通量和测序速度,同时保持了高准确性。在启动人类基因组计划时,预计要花费30亿美金、历经15年才能完成,而二代测序技术可在一个星期内完成,仅需1000美元。Solexa技术和454技术是基于连合成边测序的原理,而SOLiD技术是基于边连接边测序和双色法的原理。如前所述,二代测序技术的优点非常明显,但其缺点是在PCR扩增中增加了测序的错误率,具有明显的系统偏向性,读长较短(早期仅70多bp,最新技术也只有200多bp)。其中,读长较短给基因组的拼接带来了困难,虽然开发了大量的生物信息学算法用于二代基因组数据的拼接,但对于高杂合物种,仍然没有满意的解决途径,而绝大部分昆虫具有高杂合性。二代测序技术目前仍是市场上的主流技术,其中Illunima公司的Solexa技术因其技术优势占据了市场的半壁江山。
技术的进步是无止境的。近年来,测序技术又有了新的突破,其中主要以PacBio公司的SMRT和Oxford Nanopore Technologies公司的纳米孔单分子测序技术为代表,被称为第三代测序技术。第三代测序技术的特点是单分子测序,无需进行PCR扩增,能有效避免因PCR偏好性而导致的系统误差,同时显著提高了读长,并保持了二代测序技术高通量的优点。虽然三代测序技术已经开始走向了市场,但其准确性仍然有待高。
科研人员产生数据的能力明显地增强,海量生物数据不断积累,因此对数据管理和分析提出了更高的要求,生物信息学即在此基础上诞生。当时生物学家第一次面临超出想象的基因组数据,有点无所适从,不知所措,生物信息学俨然以“救世主”身份拯救了人类基因组计划。最被广泛接受的生物信息学定义是,综合利用生物学、计算机科学和信息科学等多学科的理论与技术,产生和创造生物数据,管理和存储生物数据,以及挖掘和分析生物数据,揭示生物数据蕴含的生物学意义。近年来,生物信息学得到了空前的充分
发展,并被不断普及。早期的生物信息研究和算法开发主要针对普遍存在的科学问题,而现在各种衍生的生物信息学算法和软件层出不穷,针对单个学科的具体科学问题进行了优化和提高,这极大地带动了大数据时代的生物信息学研究 (Ouzounisetal., 2003)。
依据研究方向,生物信息学可分为3个主要部分:(1)研发有效利用和管理数据的新工具,构建新平台,例如构建各种各样的生物信息学数据库;(2)新算法的开发,例如各类基因组测序数据的拼接和比对算法等;(3)生物数据的挖掘与分析,从海量生物数据中挖掘和发现规律,帮助生物学家从“大海捞针”变为“池塘捞鱼”,为揭示生物表型的分子机制提供有益的参考。前两个研究方向偏“信息”,而第三个研究方向偏“生物”,这与计算机科学的“偏硬”和“偏软”两个方向有异曲同工之处。生物学家更加熟悉和倚重”生物数据挖掘与分析”这一方向。但必须强调的是,数据平台和算法开发是生物信息学重要的基础,没有准确的数据,没有合适的算法,生物学意义的挖掘就无从谈起,甚至会被引至错误的方向。
2 昆虫基因组拼接、注释与数据分析
2.1 昆虫基因组组装
基因组鸟枪法是将DNA随机打断成较短的序列,构建测序载体进行测序,获得了大量的小片段序列。因此,基因组组装是基因组测序中最为关键的一步。尤其困难的是,基因组组装算法需要根据测序平台、文库构建策略和测序读长等进行优化(Richardsetal., 2015)。由于测序策略的设计缺陷或优化不足,往往导致昆虫基因组拼接失败,这样的例子并不鲜见。
根据是否有参考序列,可把基因组拼接分为从头拼接(De novo assembly)和比较拼接(comparative assembly)两大类(Wajidetal., 2012)。从头组拼接指完全依赖 reads间的重叠信息拼接出基因组序列,而比较拼接综合了reads间的重叠信息和 reads在参考序列中的位置信息,相比而言,从头拼接更难更复杂。按照算法的原理,从头拼接大致可以分以下几类:第一类是overlap/layout/Consensus(OLC)法,这类组装算法有CABOG、Newbler、Shorty、Edena、Celera等,其适应于读长较长的测序数据,如Sanger法测序和第三代测序技术,果蝇基因组的组装采用的就是Celera软件;第二类是De Bruijn Graph (DBG)法,一种基于图论的算法,软件有SOAPdenovo、Euler、Velvet等,这类算法需要不断调整k-mer的值来达到一个最佳的组装效果;第三类是Greey graph alogorithms法,这类算法有SSAKE、SHARCGS、VCAKE等(Wajidetal., 2012)。
已发表的昆虫基因组组装算法主要使用了CABOG(Milleretal., 2008)、SOAPdenove(Luoetal., 2012)、ALLPATH-LG(Butleretal., 2008)、ABySS(Simpsonetal., 2009)等方法。SOAPdenove是华大基因开发的基因短序列拼接,运行速度快,依赖于搜索k-mer来寻求最优解。ALLPATH-LG近年来使用率越来越高,特别适合于读长100-200 bp、覆盖倍数200X左右的测序策略。和SOAPdenove比,不需要设定K-mer值。但是由于其依赖穷举法,因此对硬件要求很高,运行时间非常长。
生物信息学发展至今,不断诞生了新的软件。然而,基因组组装一直都面临着巨大的挑战,无法取得理想的效果。分析认为,影响昆虫基因组拼接质量的主要原因有,一是重复序列,基因组中含有大量的重复序列,对拼接造成非常大的干扰,而昆虫基因组有可能产生了大量新的重复序列,产生了明显的影响;二是物种杂合度,当来自父本或母本染色体DNA之间的差异大时,后代可能具有更大的环境适应性优势,但给拼接造成了困难。昆虫基因组拼接困难的解决,一方面依赖于测序技术的继续进步,另一方面也依赖于算法的不断优化和提高。
2.2 昆虫基因组质量评估
目前,主要从完整性、正确性、拼接长度等几个方面进行基因组组装结果的评价(Wajidetal., 2012)。
(1)组装序列的完整性
组装序列的完整性指组装得到的基因组大小与实际基因组大小之间的差异,通常采用两者的比值来衡量。检测基因组大小的常用方法有流式细胞仪技术和K-mer分析法。
(2)拼接正确性
拼接正确性反应了组装结果和真实基因组的一致性。通常采用已知大片段序列来检测组装结果的正确性。如果没有大片段序列,可把paired-end或者mate-pair序列比对到组装结果上,检查序列在组装上的位置以及两者间的距离,以此评估拼接正确性。
(3)N50
N50是衡量基因组拼接质量的重要标准,其计算方法是,把所有序列按照从长到短进行排序,并对序列长度进行累加,当累加值达到基因组序列总数的一半时所对应的序列长度即为N50。通过计算组装基因组的contigs和scaffolds 的N50,可以非常直观的评价拼接质量。
(4)CEGMA评估
CEGMA(Parraetal., 2007)是目前使用最广泛的评估基因组甚至是转录组拼接质量的方法,其首先确定了真核生物中极其保守的248个核心基因(CEG),然后在基因组Scaffold序列中搜寻这些CEG基因,计算具有全长序列的CEG百分比、仅有部分片段的CEG百分比和完全缺失的CEG百分比,以此来判断基因组的拼接质量。
(5)BUSCO评估
BUSCO(Simaoetal., 2015)是在CEGMA上进行更新的新算法。BUSCO的其本原理与CEGMA类似并进行了优化,其按照不同的大类群选取不同的直系同源基因集,在节肢动物中挑选了2647个直系同源基因,通过检索缺失率来反映基因组质量。
2.3 昆虫基因组的注释
基因组注释是指对基因组特征进行描述,包括结构注释和功能注释。结构注释主要包括预测基因组重复序列、非编码RNA和蛋白编码基因;功能注释是根据基因序列信息预测基因的功能。
(1)重复序列注释
重复序列识别方法分为序列比对和从头预测两大类。序列比对法是根据相似性程度在基因组中识别同源的重复序列。该方法预测的结果往往比较可靠,但不全面。目前广泛使用的比对预测软件有Repeatmasker(Tarailo-Graovacetal., 2009)。从头预测方法利用重复序列的结构特征在基因组中进行预测,这种方法对结构特征明确的重复序列具有非常好的预测效果,比如MITEs、LTR等,常见的从头预测方法有Recon(Baoetal., 2002),Piler(Edgaretal., 2005),Repeatscout(Priceetal., 2005),LTR-finder(Xuetal., 2007)等。一般而言,采用同源比对和从头预测两者相结合的方法进行重复序列识别,比较可靠全面(刘金定, 2014)。
(2)非编码RNA的识别
非编码RNA指不生成蛋白产物、以RNA形式发挥功能的RNA基因,如tRNA、rRNA、piRNA、miRNA、snoRNA、rasiRNA等。非编码RNA没有蛋白质编码基因的典型特征,因此一般对其二级结构序列和特征进行预测,常用的软件有miRdeep(Friedlanderetal., 2008)、RNAstructure(Bellaousovetal., 2013)、TripletSVM(Xueetal., 2005)等,常用的非编码RNA 数据库有RNAdb(Pangetal., 2007)、NONCODE(Zhaoetal., 2016)、Rfam、miRBase(Kozomaraetal., 2014)和snoRNABase等(陈勇等, 2014)。
(3)编码基因组注释
蛋白编码基因的识别是基因组注释中最为重要的部分。常见的编码基因预测方法有基于基因模型的从头预测方法、基于比对的蛋白同源预测方法以及基于转录组比对的表达证据方法等。这3类方法各有优点和缺点:从头预测方法理论上可以覆盖全面基因集,但假阳性高;同源比对方法预测结果准确,但局限于物种间保守基因;转录组比对方法直接来自表达证据,但受限于转录组的数据质量和数量。研究人员通过整合多种预测结果来提高编码基因注释的准确性,比如Glean(Elsiketal., 2007)、Evigan(Liuetal., 2008)、PASA(Xuetal., 2006)、MAKER(Cantareletal., 2008)、jigsaw(Allenetal., 2006)等。虽然多证据整合方法可以提高编码基因注释可靠性,但是仍然也存在一些问题需要解决,比如新测序物种缺少必要数量的可靠基因用于从头预测软件训练,难以获得足够的表达证据等。真核生物广泛存在可变剪接和多个转录起始位点,导致编码基因预测更加复杂。
(4)功能注释
基因组功能注释是依据“序列决定结构,结构决定功能”的基本原理,利用序列相似性来推断基因的功能。基因功能预测是利用序列同源比对软件如Blast等搜索序列相似的已知基因,再利用已知基因的功能进行注释。常用于基因功能注释的基因集有NCBI的非冗余蛋白序列数据库(Non-redundant protein sequences, NR)、参考蛋白数据库(refseq protein)、SWISS-PROT数据库等,这些数据库中蛋白序列一般都带有注释信息。
2.4 比较昆虫基因组分析
比较基因组学是对近缘物种和同一物种的不同个体的基因组序列,从基因结构、共线性及基因家族等方面进行分析,揭示不同物种之间的基因家族扩增与丢失、基因的起源及进化等,协助阐明重要性状的分子机制。比较基因组可分为种间比较基因组和种内比较基因组,种间比较基因组是近缘物种之间的基因组比较,重点研究基因家族和基因进化;种内比较基因组比较的是同一个物种之间不同个体的遗传差异性,通过将重测序序列与参考基因组序列进行比较后,进行关联性分析,挖掘可能与重要性状关联的单核苷酸多态性和结构差异,为分子机制研究奠定基础(陈勇等, 2014)。
2.5 直系同源和共线性分析
直系同源基因具有相似的生物学功能,确定直系同源基因是功能基因鉴定、比较基因组、功能基因分类、信号通路预测等的基础。预测直系同源基因的方法大致可分为3类: 一是比较序列相似性来识别直系同源基因;二是通过构建系统发育树来识别直系同源关系;三是混合利用序列相似性和系统发育树的方法。
基因共线性(synteny)是指基因在染色体上排列顺序的一致性。在进化过程中,由于转座、插入、染色体重排、区段加倍和缺失等原因,会发现基因序列的重排,进化距离越远的物种,基因共线性越差。通过比较物种间同源基因的相对位置,可以确定不同物种间基因组的共线性,揭示所比较物种间基因结构以及基因顺序的异同。
2.6 基因家族的扩张和收缩
基因家族是来源于同一个祖先,由一个基因通过基因重复而产生两个或更多的拷贝而构成的一组基因,它们在结构和功能上具有明显的相似性,编码相似的蛋白质产物,同一家族基因可以紧密排列在一起,形成一个基因簇(gene cluster)。但多数时候,它们分散在同一染色体的不同位置,或者分布于不同染色体上,各自具有不同的表达调控模式。在长期进化过程中,基因家族会有扩张和收缩,这通常与物种的性状密切相关。
3 昆虫基因组测序计划及其进展
3.1 i5k计划
i5k计划由Gene Robinson等人(2011)在Science上发文提出,倡议在2020年前后完成5000种节肢动物基因组的测序和分析工作,建议选定的物种应该广泛分布于各种生态系统,对世界范围的农业、食品安全、药物研究、能源再生、模式生物研究等有着非常重要的影响,能够作为昆虫分类各分支上的代表物种,有助于全面理解节肢动物的进化历程和系统发育关系。我国昆虫学者积极响应i5k全球性计划,以我国昆虫学者为主导,先后完成了家蚕、小菜蛾、蝗虫、褐飞虱、榕小蜂、二化螟等昆虫的基因组测序。迄今已经召开了两届国际昆虫基因组学学术会议,分别为2013年12月15日在中国科学院动物研究所举办了“首届中国昆虫基因组学及国际i5k计划研讨会”,及于2015年9月18日在重庆召开了“第二届国际昆虫基因大会”,从基因组测序、功能基因组学、比较和进化基因组学、生物信息学技术等多个方面讨论了昆虫基因组学的发展及发展趋势,探讨了基因组学在害虫防治、资源昆虫利用、药物靶点开发及进化生物学等方面的应用前景。
3.2 已经完成的昆虫基因组测序
截至2016年11月1日,从美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI) BioProject数据库统计,共有494种昆虫的基因组测序项目在开展,覆盖了几乎所有目的昆虫。在这些的基因组测序项目中,有215个基因组完成组装并且数据已经提交到NCBI数据库,占总提交昆虫基因组测序项目的43.5%。这些物种共涵盖了15目的昆虫(图2A),包括捻翅目Strepsiptera、蜻蜓目Odonata、蜚蠊目Blattodea、直翅目Orthoptera、毛翅目Trichoptera、虱目Phthiraptera、缨翅目Thysanoptera、襀翅目Plecoptera、等翅目Isoptera、内华达古白蚁Zootermopsisnevadensis,蜉蝣目Ephemeroptera、鞘翅目Coleoptera、半翅目Hemiptera、鳞翅目Lepidoptera、膜翅目Hymenoptera和双翅目Diptera(表1)。从目的分布来看,47.17%的物种为双翅目昆虫(达100种),膜翅目占21.86%,鳞翅目占11.63%,半翅目占9.30%,鞘翅目占4.18%,其他目仅有1-2种昆虫。在双翅目昆虫中,主要为模式昆虫黑腹果蝇及其近缘种,医学昆虫蚊子等;在膜翅目昆虫中,主要为蚂蚁、蜂等;鳞翅目昆虫主要为重要农业害虫和蝶类。其中,果蝇、蚊子、蚂蚁等三类昆虫占70%以上,表明目前昆虫基因组测序仍主要为模式生物和医学昆虫等。
图2B显示了215种昆虫基因组完成测序或提交序列的时间。统计结果表明,2002-2010年期间的昆虫基因组测序进展缓慢。2010年后,在二代测序技术带动下,昆虫基因组测序的物种数大幅增长,这些“旧时王谢堂前燕”,已经“飞入了寻常百姓家”,不再是“高门槛”的项目,越来越多的实验室独立开展了昆虫基因组测序分析(张传溪, 2015)。
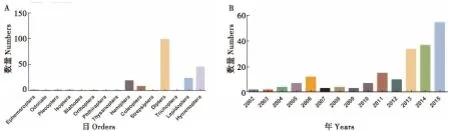
图2 已发布昆虫基因组统计Fig.2 The statistics of insect genomes have been released
从昆虫基因组数据分析来看,由于早期基因组测序是一项艰难的任务,需要庞大的人力和财力投入,基因组工作多限于数据的获得和初步分析,为分子生物学研究提供序列数据。在早期测序物种较少的情况下,比较基因组学难以展开,基因组学数据的威力一时难以完全发挥。近年来,测序物种越来越多,比较基因组分析得以深入开展,从而发现了传统思路无法发现的规律,基因组数据得到了更加充分的挖掘,为解决重要的生物学问题提供了有力的支撑。例如,对褐飞虱基因组的研究揭示了胰岛受体基因在褐飞虱翅型分化中的调控作用。
值得注意的是,在NCBI数据库注册的昆虫数要远多于提交序列的昆虫数量,而基因组数据公开发表的数量则更少。其中最为主要的原因之一,是许多昆虫的基因组拼接质量较差,还不适宜于发表。绝大多数昆虫具有非常高的杂合度,导致无法组装出高质量的基因组,影响了基因注释和后续的基因家族分析等。
3.3 重要昆虫的基因组测序及分析
如前所述,目前公开发表的昆虫基因组文章43篇涉及物种46个,昆虫基因组测序及数据分析的思路大同小异,涉及基因组拼接、注释、基因家族分析等,但针对不同昆虫的特异性表型,不同物种的分析结果各有千秋。在此,选择了一些重要的昆虫并对其基因组测序结果进行简要介绍。
3.3.1 家蚕基因组
家蚕Bomyxmori基因组于2004年完成,是继果蝇、冈比亚按蚊之后的第3个昆虫基因组,具有历史性意义。对家蚕Dazao品系进行了全基因组鸟枪法测序,基因组大小为428.7 Mb,拼接后基因组的contig N50为12.9 kb,scaffold N50为26.9 kb,共注释了18510个基因。基因组分析结果发现,家蚕基因组中含有大量的转座子插入,导致家蚕的某些基因比果蝇中的同源基因更大。在家蚕丝腺中发现了87个神经肽激素、激素受体、激素调节相关基因。在家蚕中还发现了69个与免疫相关的基因,包括moricin、cecropins、lysozymes、hemolin、lectins、prophenoloxidases等。2008年,国际家蚕基因组联盟对家蚕基因组进行了更新,提高了测序覆盖度,基因组contig N50提高为15.5 kb,scaffold N50提高到3.7 Mb,87% 的scaffold被定位于28条染色体上,预测发现了14623个基因。对新版本的基因组进行分析,发现基因组中含大量转座子,包括LINEs和SINEs两种主要类型,分别占全基因组的14.5%和13.3%。3223个家蚕特有基因在其他昆虫和脊椎动物中没有发现同源基因。研究还发现,转运Gly、 Ala和Ser的tRNA基因数目明显多于其他氨基酸tRNA,这与蚕丝蛋白中各类氨基酸含量相一致;基因Ser1、Ser2、Ser3分别编码蚕丝的不同位置和不同结构的丝胶成分;家蚕在进化过程中通过水平基因转移从细菌中获得呋喃果糖苷酶基因,得以降解桑叶中的D-AB1、DNJ等对其他昆虫有毒的生物碱类物质,这是家蚕能够专一取食桑叶的重要原因(Xiaetal., 2004)。

表 1 已发表的昆虫基因组
续上表

物种Species基因组大小(Mb)Genomesize测序平台Sequencingplatform染色体ChromosomescaffoldsN50(Kb)基因数Genenumber来源文献References松甲虫Dendroctonusponderosae246IlluminaHiseq818862813456GenomeBiol.,2013,14(3):R27 隧蜂Lasioglossumalbipes350Illumina431761613448GenomeBiol.,2013,14(12):R142 小菜蛾Plutellaxylostella383IlluminaHiseq2000181973718072NatureGenetics,2013,45(2):220-225 榕小蜂Ceratosolensolmsi268IlluminaHiseq20002457955813200GenomeBiol.,2013,14(12):R141 南极蠓Antarcticmidge99Illumina35899813517NatCommun,2014,54611无性生殖行军蚁Cerapachysbiroi206IlluminaHiseq20004579135026315CurrBiol.,2014,24(4):451-458家蝇Muscadomestica728Illumina2048722620165GenomeBiol.,2014,15(10):466 竹节虫Stickinsect1027Illumina1421131223083Science,2014,344(6185):738-742湿木白蚁Zootermopsisnevadensis472IlluminaHiseq20003162275114610NatCommun.,2014,53636蝗虫Locustamigratoria6300IlluminaHiseq2000-32017307NatCommun.,2014,52957褐飞虱Nilaparvatalugens1324IlluminaHiseq20004527936036723GenomeBiol.,2014,15(12):521草地贪夜蛾Spodopterafrugiperda358Illumina3724353711595Genomics,2014,104(2):134-143麦双尾蚜Diuraphisnoxia421IlluminaHiseq2000564139719097BMCGenomics,2015,16(1):429 咖啡果小蠹Hypothenemushampei163IlluminaHiseq20008684844719222Rep,2015,512525铜绿蝇Luciliacuprina458Illumina-ALLPATHS-LG462574414554NatCommun,2015,67344冬尺蠖蛾Operophterabrumata638IlluminaMiseq2580165616912GenomeBiolEvol,2015,7(8):2321-2332温带臭虫Cimexlectularius650Illumina-ALLPATHS-LG1402717214220NatCommun,2016,710165地中海实蝇Ceratitiscapitata479Illumina1806406014547GenomeBiol.,2016,17(1):192
3.3.2 蜜蜂基因组
蜜蜂Apismellifera基因组由The Honeybee Genome Sequencing Consortium团队于2006年完成。文章先后注释了六版基因组数据,将contig N50从19 kb提高到41 kb,scaffold N50从223 kb提高到362 kb。基因组大小236 Mb。基因组注释获得了10157个基因,比果蝇和库蚊少25%左右。蜜蜂基因组为AT-rich,高达到67%, 而黑腹果蝇Drosophilamelanogaster仅为58%, 库蚊仅为56%。在蜜蜂基因组AT丰富区中,基因分布反而较多,这与脊椎动物明显不同。蜜蜂基因组中的转座子明显比其他昆虫更少。蜜蜂和果蝇只有10%同源基因,远少于人和鸡之间有85%同源基因的比例, 表明昆虫的进化速度很快。蜜蜂有163个气味受体基因,远多于果蝇(62)和库蚊(79),显示蜜蜂化学感受能力增强, 用来探测外激素、辨别同伴和花香等。与此相反,蜜蜂的味觉基因只有10个,少于其他昆虫的50-76个。与预期相反,蜜蜂免疫和抗病基因明显变少,只有71个与免疫和抗病相关的基因,远少于库蚊的209 和果蝇的196个,分析认为这与蜜蜂的清洁行为、蜂王浆和蜂胶的抗细菌特性, 以及蜂群像城堡一样的结构等有关。研究还发现,与果蝇不同,蜜蜂有完整的DNA甲基化酶系,包括Dnmt1、Dnmt2和Dnmt3,DNA甲基化在蜜蜂不同蜂型的分化中具有重要的功能(Consortium, 2006)。
3.3.3 体虱基因组
体虱Pediculushumanus基因组于2010年完成,其基因组大小仅为108 Mb,拼接获得的基因组scaffold N50为488 kb。预测发现了10773个蛋白编码基因和57个microRNAs。与其他昆虫基因组相比,体虱具有更少的与环境感知和响应相关的基因,包括那些嗅觉和味觉感受器以及解毒酶编码的基因等。同时,还对体虱Riesia菌的基因组进行了测序。Riesia菌是体虱消化道中的一种关键细菌,它分泌营养物质作为人血的补充物质,Riesia细菌缺乏抵抗抗生素的基因。比较基因组学分析显示,人类体虱是从头虱进化而来的,基因组分析有助于利用体虱的独特基因属性如其有限的嗅觉能力等,开发出体虱控制的新方法(Kirknessetal., 2010)。
3.3.4 豌豆蚜基因组
豌豆蚜Acyrthosiphonpisum由国际蚜虫基因组联盟于2010年完成。作者利用单个雌虫的个体后代进行测序,流式细胞仪估测基因组大小为517 Mb,测序组装获得的基因组为464 Mb,基因组contig N50为10.8 kb,scaffold N50为88.5 kb,注释获得了34604个基因,远多于其他昆虫的15000-20000个,其中2459个基因家族中发现大量的基因复制,等义距离评估表明在该物种形成初期已经存在了基因复制现象,涉及功能包括染色质修饰、miRNA合成和糖转运等。豌豆蚜基因组丢失了IMD(免疫缺陷)免疫通路、硒蛋白利用、嘌呤补救途径及鸟氨酸循环等通路的基因。通过与蚜虫初级内共生菌Buchneraaphidicola基因组比较分析,发现两者具有代谢系统的互补性。豌豆蚜基因组中具有明显的基因横向转移现象,部分基因与细菌基因具有共同起源,其线粒体基因亦有部分在基因组中重复。基因组中发现了12个新的dynamin基因,可能与病毒运输、转胞等过程相关。豌豆蚜基因组中胚胎发育相关基因存在特异性的缺失,可能与其发育多型性有关。基因组中锌指结构蛋白的扩增,以及保幼激素合成酶、降解酶的hexamerin的缺失可能与豌豆蚜发育可塑性有关。
3.3.5 丽蝇蛹集金小蜂基因组
丽蝇蛹集金小蜂Nasoniavitripennis是双翅目蝇类的重要寄生蜂,其基因组测序完成于2010年。 作者采用了Sanger测序法获得26605条contigs (N50=18.5 kb),6181条Scaffolds(N50=709 kb),基因组大小约295 Mb。同时对另两种近缘寄生蜂N.giraultiandN.longicornis采用了Sanger测序技术和Illumina测序平台进行测序,得用N.vitripennis基因组做为参考,分别有62% and 62.6%的reads比对到N.vitripennis基因组上,有84.7% 和86.3%的蛋白编码区域。在N.vitripennis基因组中,注释到17279个基因,并预测了52个miRNA基因。研究发现,金小蜂具有完整的DNA甲基化“工具包”,即含有三种DNA甲基化基因,并且Dnmt1具有3个拷贝。N.vitripennis基因组的Toll通路中发现大量的基因复制。在N.vitripennis基因组中,性别决定相关基因如yellow/major、royal、jelly基因等,表现出大量的复制;N.vitripennis基因组具有与细菌Wolbachia基因相似的保守域,表明细菌基因被整合宿主基因组中,发生了基因转移现象;丽蝇蛹集金小蜂的毒液蛋白基因受到很高的进化压力。作者分析还发现,3种金小蜂线粒体基因在不同的世代受到了比较显著的进化压力(Werrenetal., 2010)。
3.3.6 帝王蝶基因组
帝王蝶Danausplexippus基因组于2011年完成,是目前唯一一篇发表于Cell杂志的昆虫基因组。帝王蝶具有迁徙和不迁徙两种类型,最早起源于美国南部和墨西哥北部的是迁徙型,大约两万年前数量增长开始迁移,向南进入南美,直到近期北美类群又分为跨太平洋和跨大西洋两个方向分布于全球各地。作者利用二代测序平台通过全基因组鸟枪法测序得到了14.7 Gb的Illumina reads,经拼接得到了273 Mb的帝王蝶基因组,注释发现了16866个蛋白编码基因。对12种昆虫和2种哺乳动物基因组进行了同源分析,结果表明鳞翅目是目前为止进化最快的昆虫;帝王蝶和家蚕在直系同源数量、微共线性、蛋白家族大小等方面具有明显的相似性。通过对帝王蝶基因组的分析,更深入地破解了其迁飞的分子机制。在帝王蝶基因组中发现了可能与处理光信号和太阳罗盘结构有关的多种蛋白和神经递质,并注释了39个与定位功能相关的基因,其中2个功能未知的基因可能是帝王蝶特有的。位于帝王蝶触角的生物钟在迁徙活动中具有重要作用,分析发现帝王蝶除了具有大量和果蝇相同的生物钟关键基因外,还具有CRY2基因,而果蝇只含有CRY1基因。保幼激素的生物合成在帝王蝶雌雄中具有两态性,表现为在雌性上调、雄性下调。研究还发现,miR-1、miR-7、miR-14在内的27种miRNA在迁徙和非迁徙蝴蝶中的表达量有差异,可能对迁飞起调节作用。独特的P型钠钾泵构成了帝王蝶防御机制的分子基础,而Ors、Grs、IRs等化学感受器在迁飞过程中也有潜在的作用。
此后,该团队采集了不同地区的101个帝王蝶基因组进行了重测序分析。在与迁徙相关的5 Mb序列中,有大约21 kb的异常序列,这段序列包含3个基因,其中Collagen IV α-1在迁徙和非迁徙群体之间具有明显的不同,从而影响了2种类型蝴蝶的体型、飞行肌以及飞行特点的不同。相比之下,迁徙蝴蝶飞行代谢率低,飞行效率高;高代谢率更有利于非迁徙蝴蝶的生存。帝王蝶特有的警戒色被发现与肌球蛋白基因DPOGS206617有密切关系,表明翅色并非由色素分子的产生决定而是由色素的运输来决定(Zhanetal., 2011)。
3.3.7 小菜蛾基因组
小菜蛾Plutellaxylostella是世界性的重要害虫,食性广,危害严重,容易对农药形成抗性,基因组大小仅为343 Mb,但其杂合度高,导致测序困难,其基因组于2013年完成测序,是第一个成功测序的高杂合度昆虫基因组。作者利用Illumina Genome Analyzer IIx和HiSeq2000平台,采用Fosmid-to-Fosmid结合WGS的测序策略,最终获得了1819条scaffold序列,N50为737 kb。基因组注释获得了18071个基因和781 ncRNA。比较基因组学分析发现,小菜蛾基因组中有1412个特有基因,参与感知和解毒代谢的基因家族发生了明显的扩张。基因组数据分析发现了在幼虫阶段偏好表达的354个基因,部分基因参与硫酸盐代谢及硫酸酯酶修饰因子基因。其中,硫代葡萄糖苷硫酸酯酶(GSS)通过催化硫代葡萄糖苷防御化合物转化为脱硫葡萄糖苷酸酯,使得小菜蛾能够在广泛的十字花科植物上进食,从而防止毒性水解产物的形成。分析认为,小菜蛾硫代葡萄糖苷硫酸酯酶(GSS)基因和硫酸酯酶修饰因子基因1(SUMF1)在幼虫时期的协同表达是决定小菜蛾能够取食十字花科蔬菜的关键。除细胞色素 (P450)、谷胱甘肽转移酶(GST)和羧基酯酶(COE)这三大代谢水解酶家族外,ABC转运蛋白家族也出现了明显的扩张,进一步解释了小菜蛾容易产生抗性的基因组学特性(Youetal., 2013)。
3.3.8 榕小蜂基因组
榕小蜂Ceratosolensolmsi在长期进化过程中,与榕属植物形成了一种密切的共生关系,是榕属植物重要的传粉媒介,以回报榕属植物为其提供栖身场所和营养来源。榕小蜂基因组于2013年完成测序和发表,其基因组大小278 Mb,scaffold数量7397。值得一提的是由于其基因组中富含AT(69.6%),重复序列只有9.85%,因此组装完成后scaffold N50值竞达到9.558 Mb,是目前测序昆虫中最高的。通过从头预测、同源搜索、转录组覆盖等方法,共注释获得蛋白质编码基因11412个。
通过比较基因组分析,发现榕小蜂的基因组进化相比于其他昆虫更快。由于榕小蜂基本上大部分时间都栖息在榕树,其基因组中ORs、GRs、IR、OBPs、CSPs等化学感受基因家族出现明显的收缩。由于榕树已为榕小蜂提供了安全的场所和营养来源,因此其P450s、GSTs、CCEs等解毒代谢基因家族基因也明显减少,以及在Toll、imd、JAK/STAT、JNK等免疫通路中很多基因退化。为了了解榕小蜂雌雄异型的分子机制,通过转录组测序技术研究了其雌雄个体中基因的表达情况,发现了很多与基因在雌雄个体中出现差异表达,推测与其这种两性差异有关。榕小蜂在长期与肠道共生菌协同进化过程中,通过基因组数据证实其可以从细菌和病毒中获得一些基因片段或完整基因,总共在榕小蜂基因组鉴定出12个水平转移基因(Xiaoetal., 2013)。
3.3.9 蝗虫基因组
蝗虫Locustamigratoria是世界范围的具有严重危害性的昆虫,其周期性的大爆发,具有长距离迁飞和两型变化的习性。蝗虫基因组达6.52 Gb,是迄今为止最大的昆虫基因组,因此完成测序极其困难,来自中国科学院动物所康乐院士所带领的团队于2014年首次解开了蝗虫的遗传密码,破解了这一难题。蝗虫基因组scaffold N50为323 kb,通过从头预测、同源预测以及表达证据共获得17307个蛋白质编码基因。基因组分析发现,蝗虫的基因组之所以如此之大,主要体现在重复序列增多,占基因组60%以上,蝗虫基因内含子的长度是其他昆虫的10倍左右,这也是造成其基因组变大的一个重要因素。
通过比较基因组学研究,发现了大量与变态发育相关的调控基因,蝗虫进化获得了55个新的基因家族,共有25个基因家族显著扩增,参与解毒代谢、化学感受、营养代谢等。蝗虫具有Dnmt1两个以及Dnmt2和Dnmt3完整的DNA甲基化基因家族,基因组中约有1.6%的胞嘧啶被甲基化,重复序列区高度甲基化。与基他昆虫不同的是,基因内含子区甲基化高于外显子区。为了适应长距离迁飞,蝗虫进化出一套高效的能量储存和代谢的机制,其主要能源物质为脂类,基因组中与脂类运输和抗氧化保护以及脂质降解有关的基因家族显著扩增,如基因组中perilipins、fatty-acid-bindingprotein、Prdx6s、sigmaGST、enoyl-CoAhydratase、acetyl-CoAacyltransferase2等基因出现多拷贝。蝗虫基因组中OBPs、ORs、GRs、IRs等基因家族出现显著的扩增,可能与其食性很广有关,同时UGTs和carboxyl/cholineesterases基因家族也出现显著扩增,以帮助其降解不同食物中的化学成分。
3.3.10 家蝇基因组
家蝇Muscadomestica是生活中常见的昆虫,幼虫以动物排泄物等为食,成虫能够携带100多种病原菌,对人类和动物的健康带来极大的威胁,其基因组测序于2014年完成。家蝇基因组大小691 Mb,重复序列含量较高,Scaffold数为20487,N50值为226 kb,基因组注释获得蛋白质编码基因14180个。在家蝇基因组中共发现771与免疫相关的基因,具有完整的Toll、imd、JAK/STAT和JNK免疫通路,这与家蝇长期生活在富含动物病原体腐烂性环境有关。先后从基因组找到146个P450s、11个P450 pseudogenes、33个GSTs、92个脂酶基因,显示家蝇基因组中解毒代谢相关的基因家族出现了明显扩张,以应对生境中各种有害物质。家蝇基因组中CysLGIC超基因家族具有23个基因,为抗药性研究和农药新靶点开发提供了参考。家蝇的味觉受体基因家族显著出现扩增,推测与家蝇需要通过味觉来识别不同的有害物质有关(Scottetal., 2014)。
3.3.11 南极蠓基因组
南极蠓Belgicaantarctica是唯一生活在南极的一种地方性昆虫,需要适应极端温度、结冰、脱水、渗透压平衡、紫外线辐射以及环境产生的其他各种选择压力,其基因组测序于2014年完成。南极蠓基因组大小89.6 Mb,是目前最小的昆虫基因组。其Contig序列为5003条,N50值为98.2 kb。虽然拼接质量不高,CEGMA基因组评估和比较基因组学研究表明南极蠓的基因组数据可以用于后续数据分析,预测得到蛋白质编码基因13517个。相比于其他昆虫,重复序列含量的大幅减少,内含子长度变短,这是其南极蠓基因组明显变小的主要原因。通过基因组个体杂合度分析发现,由于其基因组比较小,南极蠓受到的选择压力非常大,因此杂合度相对其他昆虫低。基因家族分析显示南极蠓OBP基因出现明显的收缩,推测与其生活环境、食物相对单一,活动范围也较小等习性有关(Kelleyetal., 2014)。
3.3.12 褐飞虱基因组
褐飞虱Nilapavatalugens是水稻上的重要害虫,具有迁飞习性和翅二型现象,其基因组测序完成于2014年。作者采用HiSeq2000测序技术,利用单对交配纯化13代的褐飞虱,使用与小菜蛾相似的测序策略,得到了共1.14 Gb的褐飞虱基因组序列,基因组Scaffold N50为356.6 kb,注释得到27571个蛋白编码基因。通过对褐飞虱和其它14个节肢动物基因组的比较分析,发现褐飞虱等半翅目的3个物种基因数目、特异基因数目都比其他昆虫多,显示出半翅目物种的基因扩张现象。 褐飞虱的OR和GR基因家族收缩,这与褐飞虱只以水稻韧皮汁液为食的严格单食性特性相符;研究还发现褐飞虱中解毒和消化相关基因存在着基因丢失现象,如P450、GST基因数目很少,淀粉降解必须的alpha-淀粉酶缺失,几丁质合成酶CHS2缺失,这些特点也可能与褐飞虱专一食性有关;褐飞虱与真菌YLS和细菌A.nilaparvatae组成了共生系统,通过对真菌YLS和细菌A.nilaparvatae测序并组装注释,分析三者的共生关系,发现褐飞虱缺少10种必需氨基酸合成能力,而在YLS中能找到对应的氨基酸合成基因;还发现YLS能够利用尿酸,跟褐飞虱共同形成了氮素循环的完整途径;YLS能合成酵母甾醇中间产物,褐飞虱参与利用酵母甾醇中间产物进一步合成胆固醇,从而形成完整的胆固醇合成途径;YLS和褐飞虱在维生素生物合成途径上都有缺陷,但A.nilaparvatae带有完整的维生素B合成途径,可能为褐飞虱提供维生素(Xue,etal., 2014)。
3.3.13 臭虫基因组
臭虫Cimexlectularius是与人类健康密切相关的皮外寄生物,其基因组于2016年完成。作者首先臭虫对经过6代近交纯化,然后采用二代Illumina Solexa平台测序,基因组大小为650.47 Mb,拼接得到1402条scaffold序列, scaffold N50为7.17 Mb,MAKER软件预测和手工注释共获得14220个蛋白质编码基因。基因组分析表明,为了适应臭虫独特的生态环境和生活习性,很多基因或基因家族出现了丢失或扩张。与臭虫专性寄生习性相关,在黑暗环境生存使得CRY1与JET感光基因退化,气味受体、味觉受体、离子受体等化学感受基因以及免疫通路相关基因均出现了显著的基因家族收缩;臭虫的专性吸血习性使得其唾液蛋白家族扩增,以阻止在吸食过程中的寄主血液凝固,水通道蛋白(AQP)的扩增可以快速去除血液中大量的水分;臭虫具有皮下受精交配习性,在基因组中节肢弹性蛋白基因大量扩增,使得雌虫可以最大限度地免于交配产生的创伤或修复创伤。臭虫抗药性发展迅速,基因组分析发现臭虫的电压门控钠通道基因出现了多个点突变使得靶标不敏感;差异表达分析发现P450、羧酸酯酶、谷胱甘肽-S-转移酶等代谢酶基因的表达增强,ABC转运蛋白基因家族扩增,CPR家族基因扩增等均是造成了臭虫日趋严重抗性的原因。通过微生物和寄主分析,发现了臭虫与其体内walbacia菌形成营养共生关系,在臭虫基因组发现了805个潜在的水平转移基因。臭虫基因组使得从分子机制水平研究和解释臭虫的寄生习性、嗜血习性、抗药性等科学问题成为可能,为研究吸血昆虫、共生关系以及寄生行为等提供了新的模式材料(Benoit,etal., 2016)。
3.3.14 地中海实蝇
地中海实蝇Ceratitiscapitata是世界性的入侵害虫,其基因组大小为479 Mb,基因组测序完成于2016年。作者先后采用454平台和Illumina HiSeq2000平台进行测序,利用单对纯化后的个体DNA进行测序以提高数据质量,将contig N50从3.1 kb提高到45.8 kb,Scaffold N50从29.4 kb提高到4.1 Mb。基因组注释获得14547个基因,23075个CDS。与其它14个节肢动物的基因组进行同源分析,确定了26212个同源组。地中海实蝇中有1608条推定的氨基酸序列没有分到任何同源组内,推测是最近才进化的新基因。利用地中海实蝇的唾液腺多线染色体,通过克隆基因和微卫星序列(Medflymic)的原位杂交,将克隆基因和微卫星序列所在的43个scaffold定位到5条常染色体上(染色体2-6号),1个scaffold定位到X性染色体上。与黑腹果蝇和家蝇基因组进行比较分析,发现多个基因/基因家族的扩张现象可能导致地中海实蝇较高的适应性和入侵性,包括IR和GR味觉受体基因家族、性诱剂受体、细胞色素P450基因和CYP6亚家族、免疫系统基因(Toll和spätzle家族)、TWDL和CPLCA表皮蛋白家族、水通道蛋白基因以及特异的ceratotoxin基因。对各基因家族的分析表明,可利用化学感受分子作为种群监测或诱捕的引诱剂或驱避剂,视蛋白opsin指导最佳陷阱颜色的选择,RHG促细胞凋亡基因(reaper、grim)、精液蛋白SFP等用于SIT昆虫不育技术(Papanicolaouetal., 2016)。
4昆虫基因组数据库
随着测序技术的突破性发展,海量的生物数据在不断累积,每14个月就会增长一倍,如何进行数据的管理、存储、展示、共享,变成了非常迫切的问题(Baxevanisetal., 2015, Stephensetal., 2015)。为了最大化地体现数据的价值和提高数据的利用率,数据库在管理和维护、共享与挖掘生物大数据中发挥着重要作用。
依据数据资源分类,生物数据库可以分为三类。第一类是大型综合存储型数据库。这类数据库的特点就是,大而杂地收录了大量的数据,数据之间层次和质量良莠不齐,且仅仅是接近原始版的堆积,更新、修改和管理较为困难,而且数据库比较大,维护的成本很高,主要是发挥数据仓库的作用。这类数据库以美国国家生物技术信息中心(NCBI)、欧洲生物信息研究所(EBI)和日本核酸数据库(DDBJ)国际上公认的三大生物信息数据库为代表,这三个数据库各具特色。第二类是单一类群的基因组数据库。这类数据库是围绕某一个研究类群的基因组数据库,数据量较第一类数据库明显缩小,数据之间的层次和质量比较接近,且质量有所保证,数据也经过了加工,维护者管理起来也比较方便,使用者用起来也可以很快的掌握。VectorBase (Giraldo-Calderonetal., 2015)是这类型数据的经典代表,其中收录了与众多与疾病媒介传播有关物种的基因组数据。第三类是小型的单个物种或单一属的物种数据库,围绕单一物种的数据构建数据库,数据质量很高,数据加工很精细,功能很齐全,维护和更新迅速和简便,使用便捷。这类数据库目前有膜翅目数据库Hymenoptera Genome Database(Munoz-Torresetal., 2011)、农业害虫数据库Agripestbase、小菜蛾数据库(中国)DBM-DB(Tangetal., 2014)、小菜蛾数据库(日本)KONAGAbase(Jourakuetal., 2013)、帝王蝶数据库MonarchBase(Zhanetal., 2013)、蚜虫数据库APHIDBASE(Legeaietal., 2010)、家蚕数据库(中国)SilkDB(Duanetal., 2010, Wangetal., 2005)、家蚕数据库(日本)KAIKObase(Shimomuraetal., 2009)、诗神袖蝶数据库Heliconius Genome Project、二化螟数据库ChiloDB(Yinetal., 2014)和WaspAtlas金小峰数据库(Daviesetal., 2015)。
目前昆虫基因组数据主要存储于大型综合存储型数据库中。NCBI共收录了215个昆虫的基因组拼接数据,Ensemble上收录了31个,这两个公共数据库涵盖了大部分的昆虫基因组数据。由于NCBI等大型数据库并不是单一地为昆虫领域服务,主要集中在医学、模式生物领域。目前NCBI基本没有针对昆虫基因组数据进行挖掘和数据注释等,仅仅只是数据仓库服务。为此,这么多昆虫基因组研究者纷纷建立了单个类群或单个个体的基因组数据库(表2),在众多的昆虫基因组数据库,涌现了2个综合型的昆虫基因组数据库,i5k workspace@NAL(Poelchauetal., 2015)和InsectBase(Yinetal., 2016)。
4.1 i5k Workspace@NAL
i5k Workspace@NAL数据库是由美国农业部主导构建的节肢动物基因组学服务型数据库,共收录昆虫基因组46个,数据库提供基因组数据的浏览、下载、数据提交、序列比对、基因组可视化及在线基因组手工注释平台,以及HMMER、CLUSTAL两个在线工具(Poelchauetal., 2015)。随着i5k计划的提出,越来越多的节肢动物基因组被测序。在此背景下,美国农业部相关科学家希望在纷乱无章的测序潮流中推出一套基因组测序、组装、注释、维护、共享的标准化流程和平台,因此构建了i5k Workspace@NAL数据库。然而事与愿违,在目前基因组数据依旧是稀缺资源的环境下,大多数研究人员没有遵从i5k Workspace@NAL提出的共享数据标准。目前,i5k Workspace@NAL主要收录了美国农业部主导的一些节肢动物基因组测序数据,其他国家科学几乎没有提交数据。
4.2 InsectBase
InsectBase昆虫基因组与转录组数据库旨在有效的解决目前昆虫基因组数据库的纷乱杂陈的现状,构建一个综合的全能化的昆虫领域的生物信息数据库,为广大研究者提供方便快捷的后基因组时代基因组、转录组等数据服务和交流合作平台(Yinetal., 2016)。
InsectBase昆虫基因组数据库(http://www.insect-genome.com/)的总数据存储量达120 G。InsectBase通过筛选和质量过滤共收集了155种昆虫基因组(隶属于16个目),其中61个基因组具有注释信息(Official Gene Set, OGS),116个转录组数据,237个物种的EST序列,69个物种的7544条miRNA序列,2个物种的83262条piRNA序列,构建了78个物种的22536个信号通路,116个昆虫的UTR序列和CDS序列。针对61个有OGS注释的昆虫,开展了数据挖掘。
InsectBase对研究较多的36个基因家族开展了系统分析,运用OrthoMCL直系同源算法发现了7个物种中的直系同源基因,共找到1 ∶1 ∶1直系同源基因973个。InsectBase昆虫基因组数据库提供序列查询、序列比对、基因组可视化、信号通路和注释、进化分析和进化树构建等功能服务,所有基因数据均可下载。从PubMed中下载了94758条昆虫研究相关文献,通过数据挖掘,建立了昆虫学领域的关系网络平台iFacebook,初步实现“基因-研究者-昆虫物种”等三者之间的关系网络,便于促进学术交流。InsectBase是综合型的生物信息学数据库,数据种类齐全、功能全面、用户使用方便,有利于昆虫学研究者对基因数据的获得、整理和分析,促进昆虫分子生物学研究。自2015年8月上线以来,到目前已经累计有来自全世界86个国家的研究学者近10万次的访问,其中最活跃的当属中国和美国,中国的访问量占到86.23%。

表 2 昆虫基因组数据库统计
5 总结与展望
随着测序费用的急剧下降,昆虫基因组测序计划如雨后春笋般地涌现。由于昆虫基因组杂合度高导致的拼接困难等问题,在2020年前完成5000种昆虫测序的目标也许很难实现,但随着技术的进步,这些困难最终会得到彻底解决。对948种昆虫基因组大小进行统计分析,结果显示平均大小为1.15 Gb,按1000美元完成人基因组(3 Gb)测序来计算,完成一个昆虫基因组的测序仅需不到400美元。相信在不久的将来,昆虫基因组测序和重测序将成为日常实验设计的一部分。
组学数据的大量积累,将会对昆虫学研究起巨大的推动作用。首先,系统生物学的研究思路将占据昆虫分子生物学研究的高地,研究人员不仅仅将基因组作为数据仓库在使用,而且可以从组学角度寻找重要科学问题的答案,才是功能基因组学研究时代的突破性飞跃。其次,生物数据的积累对生物信息学提出了更高的要求。目前,数据分析工作主要依赖于公司的技术人员完成,但是常规的通用分析流程将越来越不能胜任具有针对性的数据分析需求,生物信息学技术将如同上世纪90年代末的分子生物学技术一样,成为每一个实验室的重要技术平台。因此,昆虫学研究中应该注重培养既懂昆虫学问题也熟悉生物信息学分析的两栖人才。最后,基因组重测序、转录组、蛋白组和代谢组等将成为功能基因组时代的四驾马车,将DNA、RNA、蛋白质和代谢产物4个不同层次的大数据充分整合,是功能基因组时代的重要研究手段。
在昆虫基因组学研究中,还应当注意和明确的是,数据和技术应该为科学问题服务。昆虫基因组数据的大量堆积,数据质量良莠不齐,需要提高和发展;技术层面上的问题重重,需要实现突破。他山之石,可以攻玉。昆虫基因组研究可以并应当借鉴医学研究领域的领先技术和思路,但技术的突破和数据的提高,应该紧密围绕昆虫科学问题,服务于害虫防治和益虫利用的最终目标。
References)
Adams MD, Celniker SE, Holt RA,etal. The genome sequence of Drosophila melanogaster [J].Science, 2000, 287(5461): 2185-95.
Allen JE, Majoros WH, Pertea M,etal. JIGSAW, GeneZilla, and GlimmerHMM: Puzzling out the features of human genes in the ENCODE regions [J].GenomeBiol., 2006, 7(S9):1-13.
Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes [J].GenomeRes., 2002, 12(8): 1269-1276.
Baxevanis AD, Bateman A. The importance of biological databases in biological discovery [J].CurrProtocBioinformatics, 2015, 50111-50118.
Bellaousov S, Reuter JS, Seetin MG,etal. RNAstructure: Web servers for RNA secondary structure prediction and analysis [J].NucleicAcidsRes., 2013, 41(Web Server issue): W471-474.
Benoit JB, Adelman ZN, Reinhardt K,etal. Unique features of a global human ectoparasite identified through sequencing of the bed bug genome [J].Nat.Commun., 2016, 710165.
Butler J, MacCallum I, Kleber M,etal. ALLPATHS: De novo assembly of whole-genome shotgun microreads [J].GenomeRes., 2008, 18(5): 810-820.
Cantarel BL, Korf I, Robb SM,etal. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes [J].GenomeRes., 2008, 18(1): 188-196.
Chen Y,Liu YS,Zeng JG,etal. Progresses on plant genome sequencing profile [J].LifeScienceResearchFeb.,2014(1): 66-74.
Consortium HGS. Insights into social insects from the genome of the honeybeeApismellifera[J].Nature, 2006, 443(7114): 931.
Davies NJ, Tauber E. WaspAtlas: A Nasonia vitripennis gene database and analysis platform [J].Database(Oxford), 2015.
Duan J, Li R, Cheng D,etal. SilkDB v2.0: A platform for silkworm (Bombyxmori)genome biology [J].NucleicAcidsRes., 2010, 38(Database issue): 453-456.
Edgar RC, Myers EW. PILER: Identification and classification of genomic repeats [J].Bioinformatics, 2005, 21(Suppl):152-158.
Elsik CG, Mackey AJ, Reese JT,etal. Creating a honey bee consensus gene set [J].GenomeBiol., 2007, 8(1): R13.
Friedlander MR, Chen W, Adamidi C,etal. Discovering microRNAs from deep sequencing data using miRDeep [J].Nat.Biotechnol., 2008, 26(4): 407-415.
Giraldo-Calderon GI, Emrich SJ, MacCallum RM,etal. VectorBase: An updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases [J].NucleicAcidsRes., 2015, 43(Database issue): 707-713.
Heather JM, ChainB. The sequence of sequencers: The history of sequencing DNA[J].Genomics, 2016, 107(1): 1-8.
Jouraku A, Yamamoto K, Kuwazaki S,etal. KONAGAbase: A genomic and transcriptomic database for the diamondback moth,Plutellaxylostella[J].BMCGenomics, 2013: 14464.
Kelley JL, Peyton JT, Fiston-Lavier AS,etal. Compact genome of the Antarctic midge is likely an adaptation to an extreme environment [J].Nat.Commun., 2014, 54611.
Kirkness EF, Haas BJ, Sun W,etal. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle [J].ProceedingsoftheNationalAcademyofSciences, 2010, 107(27): 12168-12173.
Kozomara A, Griffiths-Jones S. miRBase: Annotating high confidence microRNAs using deep sequencing data [J].NucleicAcidsRes., 2014, 42(Database issue): 68-73.
Legeai F, Shigenobu S, Gauthier JP,etal. AphidBase: A centralized bioinformatic resource for annotation of the pea aphid genome [J].InsectMol.Biol., 2010, 19(Suppl):25-12.
Liu JD,Improvement of Insect Genome Annotation Method and Analysis of Two Insect Genomes [D]. Nanjing Agricultural University,2014.
Liu Q, Mackey AJ, Roos DS,etal. Evigan: A hidden variable model for integrating gene evidence for eukaryotic gene prediction [J].Bioinformatics, 2008, 24(5): 597-605.
Luo R, Liu B, Xie Y,etal. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler [J].Gigascience, 2012, 1(1): 18.
Maxam AM, Gilbert W. A new method for sequencing DNA [J].Proc.Natl.AcadSci.USA, 1977, 74(2): 560-564.
Miller JR, Delcher AL, Koren S,etal. Aggressive assembly of pyrosequencing reads with mates [J].Bioinformatics, 2008, 24(24): 2818-2824.
Munoz-Torres MC, Reese JT, Childers CP,etal. Hymenoptera Genome Database: Integrated community resources for insect species of the order Hymenoptera [J].NucleicAcidsRes., 2011, 39(Database issue): 658-662.
Ouzounis C A, Valencia A. Early bioinformatics: The birth of a discipline—a personal view [J].Bioinformatics, 2003, 19(17): 2176-2190.
Pang KC, Stephen S, Dinger ME,etal. RNAdb 2.0—An expanded database of mammalian non-coding RNAs [J].NucleicAcidsRes., 2007, 35(Database issue): 178-182.
Papanicolaou A, Schetelig MF, Arensburger P,etal. The whole genome sequence of the Mediterranean fruit fly,Ceratitiscapitata(Wiedemann), reveals insights into the biology and adaptive evolution of a highly invasive pest species [J].GenomeBiol., 2016, 17(1): 192.
Parra G, Bradnam K, Korf I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes [J].Bioinformatics, 2007, 23(9): 1061-1067.
Poelchau M, Childers C, Moore G,etal. The i5k Workspace@NAL—enabling genomic data access, visualization and curation of arthropod genomes [J].NucleicAcidsRes, 2015, 43(Database issue): 714-719.
Price AL, JonesNC, Pevzner PA. De novo identification of repeat families in large genomes [J].Bioinformatics, 2005, 21(Suppl.):351-358.
Richards S, Murali SC. Best Practices in Insect Genome Sequencing: What Works and What Doesn’t [J].Curr.Opin.Insect.Sci., 2015, 71-77.
Robinson GE, Hackett KJ, Purcell-Miramontes M,etal. Creating a buzz about insect genomes [J].Science, 2011, 331(6023): 1386-1386.
Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase [J].J.Mol.Biol., 1975, 94(3): 441-448.
Sanger F, Air GM, Barrell BG,etal. Nucleotide sequence of bacteriophage phi X174 DNA [J].Nature, 1977, 265(5596): 687-695.
Scott JG, Warren WC, Beukeboom LW,etal. Genome of the house fly,MuscadomesticaL., a global vector of diseases with adaptations to a septic environment [J].GenomeBiol., 2014, 15(10): 466.
Shimomura M, Minami H, Suetsugu Y,etal. KAIKObase: An integrated silkworm genome database and data mining tool [J].BMCGenomics, 2009, 10486.
Simao FA, Waterhouse RM, Ioannidis P,etal. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs [J].Bioinformatics, 2015, 31(19): 3210-3212.
Simpson JT, Wong K, Jackman SD,etal. ABySS: A parallel assembler for short read sequence data [J].GenomeRes., 2009, 19(6): 1117-1123.
Stephens ZD, Lee SY, Faghri F,etal. Big Data: Astronomical or Genomical?[J].PLoSBiol., 2015, 13(7): e1002195.
Tang W, Yu L, He W,etal. DBM-DB: The diamondback moth genome database [J].Database(Oxford), 2014.
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences [J].Curr.Protoc.Bioinformatics, 2009, Chapter 4Unit 4 10.
Venter JC, Adams MD, Myers EW,etal. The sequence of the human genome [J].Science, 2001, 291(5507): 1304-1351.
Wajid B, Serpedin E. Review of general algorithmic features for genome assemblers for next generation sequencers [J].GenomicsProteomicsBioinformatics, 2012, 10(2): 58-73.
Wang J, Xia Q, He X,etal. SilkDB: A knowledgebase for silkworm biology and genomics [J].NucleicAcidsRes., 2005, 33(Database issue): 399-402.
Wang X, Fang X, Yang P,etal. The locust genome provides insight into swarm formation and long-distance flight [J].Nat.Commun., 2014: 52957.
Werren JH, Richards S, Desjardins CA,etal. Functional and evolutionary insights from the genomes of three parasitoidNasoniaspecies[J].Science, 2010, 327(5963): 343-348.
Xia Q, Zhou Z, Lu C,etal. A draft sequence for the genome of the domesticated silkworm (Bombyxmori)[J].Science, 2004, 306(5703): 1937-1940.
Xiao JH, Yue Z, Jia LY,etal. Obligate mutualism within a host drives the extreme specialization of a fig wasp genome [J].GenomeBiol., 2013, 14(12): R141.
Xu Y, Wang X, Yang J,etal. PASA—a program for automated protein NMR backbone signal assignment by pattern-filtering approach [J].J.Biomol.NMR, 2006, 34(1): 41-56.
Xu Z, Wang H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons [J].NucleicAcidsRes., 2007, 35(Web Server issue): 265-268.
Xue C, Li F, He T,etal. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine [J].BMCBioinformatics, 2005:6310.
Xue J, Zhou X, Zhang CX,etal. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation [J].GenomeBiol., 2014, 15(12): 521.
Yin C, Liu Y, Liu J,etal. ChiloDB: A genomic and transcriptome database for an important rice insect pestChilosuppressalis[J].Database(Oxford), 2014.
Yin C, Shen G, Guo D,etal. InsectBase: A resource for insect genomes and transcriptomes [J].NucleicAcidsRes., 2016, 44(D1): 801-807.
You M, Yue Z, He W,etal. A heterozygous moth genome provides insights into herbivory and detoxification [J].NatureGenetics, 2013, 45(2): 220-225.
Zhan S, Merlin C, Boore J L,etal. The monarch butterfly genome yields insights into long-distance migration [J].Cell, 2011, 147(5): 1171-1185.
Zhan S, Reppert S M. MonarchBase: The monarch butterfly genome database [J].NucleicAcidsRes., 2013, 41(Database issue): 758-763.
Zhang CX,Current research status and prospects of genomes of insects important to agriculture in China [J].ScientiaAgriculturaSinica,2015(17): 3454-3462.
Zhao Y, Li H, Fang S,etal. NONCODE 2016: An informative and valuable data source of long non-coding RNAs [J].NucleicAcidsRes., 2016, 44(D1): 203-208.
Chen Y,Liu YS,Zeng JG.Progresses on plant genome Sequencing profile[J].LifeScienceResearch,2014,18(1):66-74.[陈勇, 柳亦松, 曾建国. 植物基因组测序的研究进展[J]. 生命科学研究, 2014,18(1): 66-74]
Liu JD.Improlement of insect genome annotation method and analysis of two insect geomes[D].Nanjing Agriculture University,2014.[刘金定. 昆虫基因组注释方法改进及两种昆虫基因组分析[D].南京农业大学, 2014]
Zhang CX.Current research status and prospects of genomes of insect important to agriculture in China[J].ScientiaAgricutturaSinica,48(17):3454-3462.[张传溪. 中国农业昆虫基因组学研究概况与展望[J]. 中国农业科学, 2015,48(17): 3454-3462]
The progress of insecg genomic research and the gene database
YIN Chuan-Lin, LI Mei-Zhen, HE Kang, DING Si-Min, GUO Dian-Hao, XI Yu, LI Fei*
(Institute of Inesct Science, Zhejiang University,Hangzhou 310058, China)
With huge amount of insect genome sequencing data was generated, entomology has entered a new era of systematic biology. Up to now, 467 insect genome projects have been registered on NCBI, among which 225 have submitted with sequencing raw reads, 215 have been assemblied, 65 have been annotated and 43 have been published. Here, we reviewed the development of different sequence technologies, methods and problems of genome assembly, genome annotation and analysis, and important achievements in the field of insect genome projects. In addition, we summarized the development of insect genome databases. Insect genomics is now a hotspot of scientific study, which has wide applications in pest control and utilization of the resource insects.
Insect genome; genome database; big DATA; biological databases

特邀稿件InvitedReview
国家重点研发计划“主要入侵生物的生物学特性分析”重大课题(2016YFC1200602)
尹传林,男,1989年生,博士研究生,研究方向为昆虫基因组学,E-mail: yincl2013@126.com
*通信作者Author for correspondence, E-mail: lifei18@zju.edu.cn
Q963; S43
A
1674-0858(2017)01-0001-18
Received:2016-12-10;接收日期 Accepted:2016-12-20
尹传林,李美珍,贺康,等.昆虫基因组及数据库研究进展[J].环境昆虫学报,2017,39(1):1-18.
