Leigh综合征1例报告
2017-03-16刘雪梅魏晓晶苏飞飞段晨晨于雪凡
刘雪梅, 魏晓晶, 苏飞飞, 段晨晨, 苗 晶, 于雪凡
Leigh综合征1例报告
刘雪梅, 魏晓晶, 苏飞飞, 段晨晨, 苗 晶, 于雪凡
Leigh综合征(leigh syndrome,LS)又称亚急性坏死性脑脊髓病(syndrome subacute necrotzing encephalopathy),是一种由线粒体氧化磷酸化缺陷导致的多基因遗传的神经退行性疾病,由Leigh于1951年首次报道,是婴儿和儿童时期最常见的一种线粒体疾病,其影像学特征是基底节、丘脑、脑干和脊髓的对称性病变,呈母系遗传、常染色体隐性遗传或X-连锁遗传,具有临床表型和遗传异质性[1],故易造成漏诊或误诊。现报道1例儿童期起病且长时间存活的Leigh综合征。
1 临床资料
患者,女,37岁,因“行走不稳伴言语笨拙30 y,加重4 y,听力下降5 m”收入院。患者30 y前无明显诱因出现行走不稳,表现为步态异常、易跌倒,伴言语笨拙,表现为语速慢、吐字不清,未在意未治疗;4 y前上述症状较前加重;5 m前家人发现其听力下降,未经治疗,现为求进一步诊治来我院。病程中,伴智能减退,理解力低于同龄人,运动能力差,且进行性加重,现仅能行走数步,伴头晕,无头痛、恶心、呕吐,无视物障碍,无抽搐、意识障碍,无饮水呛咳、吞咽困难。发病来,饮食、睡眠可,二便如常,体重未见明显变化。既往:足月顺产,母乳喂养;否认感染、中毒史、脑血管病史。育有1子16岁,体健;父母非近亲结婚,父母家族无类似疾病史。入院后查体:身材矮小,体型偏瘦。神志清楚,构音障碍,表情呆滞,记忆力、计算力减退。双侧瞳孔等大等圆,直径约3.0 mm,直、间接对光反射灵敏,细小水平眼震,双眼外展稍受限,伸舌居中,双侧软腭上抬有力,悬雍垂居中,咽反射存在,双耳听力粗测下降,颈伸肌、颈屈肌4+级,耸肩有力,双上肢近端肌力4+级,远端肌力5级,双下肢肌力4级,四肢肌张力低下,双上肢腱反射正常,双下肢腱反射活跃,双侧Babinski征、Chaddock征阴性,无项强,克氏征阴性。双侧指鼻试验、跟膝胫试验欠稳准,双侧轮替试验笨拙,昂白氏征不能。余神经系统查体未见异常。辅助检查:肌酸激酶278 U/L(25~200 U/L),肌酸激酶同工酶 26.1 U/L(0.0~25 U/L),乳酸脱氢酶 302 U/L(135~226 U/L),α-羟丁酸脱氢酶 240 U/L(78.0~182.0 U/L);甲功5项:促甲状腺激素7.55 μIU/ml(0.27~4.2 μIU/ml);风湿3项、免疫5项、血沉、ANA系列、肝功、铜蓝蛋白测定、腹彩未见异常。K-F环阴性。肌电图:上下肢所测神经、肌肉均未见异常。头部MRI(见图1):双侧基底节区对称性长T1、长T2信号。肌肉活检病理(见图2):HE染色可见肌纤维大小不等,部分肌纤维膜下可见嗜碱性深染物质聚集,GT染色为RRF,NADH、SDH、COX染色可见部分肌纤维内酶活性不均匀,SDH染色可见数条蓝纤维,COX染色可见部分阴性肌纤维。
2 讨 论
Leigh综合征是一种婴儿及儿童早期的疾病,成人罕见,且多于2岁前发病。临床表现多样,从无任何异常到严重的神经系统问题,其中枢神经系统最常见的临床表现是精神运动发育迟缓、眼球震颤、眼肌麻痹、视神经萎缩、共济失调、肌张力障碍、或呼吸衰竭[2]。Leigh综合征临床表现的复杂性及临床医生对该病认识的不足,使该病正确的临床诊断有一定困难。目前Leigh综合征尚无公认的诊断标准,结合Rahman、吴斌等人[3,4]提出的Leigh综合征诊断标准主要包括:(1)典型的临床表现;(2)神经影像学特征(脑干和/或基底节对称性病变);(3)血液和/或脑脊液中乳酸水平升高;(4)肌肉活检可见异常肌纤维和/或死后的病理改变;(5)排除代谢障碍、中毒、感染等疾病。典型的神经影像学特点对于本病的诊断尤为重要。
本患者为青年女性,儿童期起病,病情进展缓慢;具有典型的临床症状,身材矮小、运动智力发育迟缓,肌张力低下、共济失调、听力下降、脑干症状(构音障碍、眼震、眼外肌麻痹);头部MRI表现为基底节区对称性长T1、长T2信号;故临床诊断为Leigh综合征。该患者属于儿童晚期发病且长时间存活的Leigh综合征,有文献报道[5]儿童晚期、青少年发病的Leigh综合征症状大多数缓慢进展,但可长时间存活,本例报道与文献相符。肌肉活检破碎红纤维是线粒体疾病的标志,但Leigh综合征罕见[5],而本例患者肌肉活检发现RRF和COX染色缺失,但有文献报道儿童型Leigh综合征ND5突变肌肉活检中可发现RRF和COX染色酶活缺失[6]。大多数Leigh综合征患者血和/或脑脊液中乳酸水平升高,但是这并不是一个绝对性标准,有研究发现25%的患者血液和/或脑脊液中乳酸正常,本报道患者未行此检验,但仍可以诊断为Leigh综合征。基因异常也有助于Leigh综合征的诊断,但仅有不到35%~65%的Leigh综合征患者可检测到基因异常[2],所以对于可疑Leigh综合征但基因检测正常的患者,应注意寻找其他证据。
目前,Leigh综合征尚无有效的治疗方法,主要改善临床症状、提高生活质量,其治疗原则主要是补充线粒体氧化呼吸链中的相关辅酶,如各种维生素、辅酶Q10、左旋肉碱等“鸡尾酒”疗法。Leigh综合征的预后与发病年龄有很大关系,常见的死亡原因是中枢性呼吸衰竭。临床上影像学表现为双侧基底节区对称性低密度(头部CT)或长T1、长T2信号(头部MRI)可见于多种疾病,主要包括代谢性疾病(肝豆状核变性、婴幼儿维生素B1缺乏、Leigh 综合征)、感染性疾病(EB病毒性脑炎)、中毒性疾病(CO中毒、变质甘蔗中毒)、血管性疾病(脑梗死、缺血缺氧性脑病后遗改变)[7、8]。若具有上述特征性影像学表现,且可排除感染、中毒、血管性疾病,应注意考虑Leigh综合征诊断的可能,详细询问病史,并尽可能完善血或脑脊液乳酸、肌肉活检、基因检测等相关检验、检查。
[1]Leigh D.Subacute necrotizing encephalomyelopathy in an infant[J].J Neurol Neurosurg Psychiatry,1951,14:216-221.
[2]Finsterer J.Leigh and leigh-like syndrome in children and adults[J].Pediatr Neurol,2008,39:223-235.
[3]Rahman S,Blok RB,Dahl H-HM,et al.Leigh syndrome:Clinical features and biochemical and DNA abnormalities[J].Ann Neurol,1996,39:343-351.
[4]Sakushima K,Tsuji-Akimoto S,Niino M,et al.Adult Leigh disease without failure to thrive[J].Neurologist,2011,17(4):222-227.
[5]Gerards M,Sallevelt SC,Smeets HJ,et al.Leigh syndrome:Resolving the clinical and genetic heterogeneity paves the way for treatment options [J].Moecular Genetics and Metabolism,2016,117:300-312.
[6]Shanske S,Coku J,Lu J,et al.The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome[J].Arch Neurol,2008,65(3):368-372.
[7]Tabarki B,Al-Shafi S,Al-Shahwan S,et al.Biotin-responsive basal ganglia disease revisited:clinical,radiologic,and geneic findings[J].Neurology,2013,80(3):261-267.
[8]吉六舟,李洪涛,孙国运,等.双侧基底节区对称性低密度病变的临床与CT分析[J].中国医学影像学杂志,2008,16(2):127-129.

A:T1WI;B:T2WI;C:FLAIR
图1 头部MRI可见双侧壳核后部对称性病变,呈长T1、长T2信号影,边界清楚
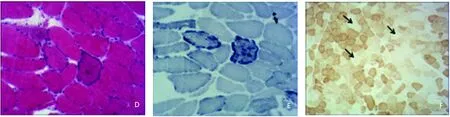
D:HE染色(×100)肌纤维大小不等,部分肌纤维膜下可见嗜碱性深染物质聚集,肌纤维膜边缘不整,呈破碎状;E:SDH染色(×100)可见数条蓝纤维;F:COX染色(×100)可见部分阴性肌纤维(箭头)
图2 肌肉活检病理
1003-2754(2017)02-0177-02
R742
2016-10-26;
2016-11-29
(吉林大学白求恩第一医院神经内科和神经科学中心,吉林 长春 130021)
于雪凡,E-mail:dr_yuxuefan@126.com
