福岛核事故后上海气溶胶中131I、134Cs和137Cs的来源途径分析
2017-03-01刘丹彤王锦龙毕倩倩杜金洲
刘丹彤,王锦龙,毕倩倩,杜金洲
福岛核事故后上海气溶胶中131I、134Cs和137Cs的来源途径分析
刘丹彤,王锦龙,毕倩倩,杜金洲*
北京时间2011年3月11日北太平洋西海岸发生的剧烈地震和海啸导致了日本福岛核电站事故。据估计,福岛核事故释放的放射性核素总量为340~800 PBq[1-2]。这些放射性物质主要经由三种通道向外界传播,即大气通道、海洋表层海流通道和海洋内部水体通道[3-4]。由于日本近海的洋流流向以及与中国海之间存在的强盛海流——黑潮(Kuroshio)的影响,放射性物质通过洋流输送至中国海近岸需要一定的时间,然而大气扩散可以使放射性物质快速输运到中国境内。因此,放射性气溶胶作为一个灵敏的核事故检测项受到了广泛地关注[5-8]。
气溶胶是指在大气中悬浮的固态和液态微粒,是大气的重要组成成分,参与了大气中的许多物理和化学过程[9]。核试验或者核事故释放的放射性物质进入大气后,有些吸附在气溶胶上,有些则被气溶胶包覆,并随之扩散。以气溶胶为载体的放射性核素的研究为综合评估气溶胶的输运和大气对不同来源物质的载荷能力提供了行之有效的手段[10]。因此,核事故期间携带有放射性核素的气溶胶的来源、输运和清除等物理、化学行为已经成为国内外学者广泛研究的课题。
Menut等[11]2004年检测到法国南部发生沙尘暴后气溶胶中的137Cs活度显著升高,并结合CHIMERE-DUST传输模型判定这场沙尘的最终来源是非洲北部;万国江等[12]对黔中气溶胶的传输进行了210Pb和7Be示踪,解释了周时间尺度的含义;樊元庆等[13]发现北京地区大气中137Cs的监测与气象条件密切相关,其主要来源是地表沉降的再悬浮,而131I的监测条件除了与气象条件有关外,还与其监测位置的131I本底值相关,因为该研究的监测位置周边存在较多贮存和使用131I的医疗机构,造成131I的监测次数和活度值显著偏高。福岛核事故发生的时间段内,受西风带影响,大部分气溶胶不会直接到达距离日本较近的韩国、中国等地,而是向东和东北方向运移,穿过太平洋上空先到达美洲[14-15];Takemura等[16]在研究中也指出,2011年3月14日—15日期间日本存在的由低压系统控制的大规模上升气流,能够有效地将地面表层的颗粒带入至西风急流系统,可以在3~4 d内使这些放射性核素横跨太平洋。
气团运移轨迹模型HYSPLIT(hybrid single-particle lagrangian trajectory) 是研究气团输送的有效工具,该模型利用气象场中四维(x,y,z,l)数据,通过计算羽流或粒子的扩散,进而实现了模拟大气中颗粒物复杂的运移、扩散和沉积过程的目的[14,17]。
本工作通过对上海地区福岛核事故期间气溶胶中131I和134, 137Cs活度及其比值分布特征规律的分析,结合中国境内相关省份监测到的大气数据,归纳得到大气中放射性物质在中国境内的迁移途径以及区域性大气放射性监测的影响因素;用HYSPLIT 模型定性验证放射性气团从福岛核电站到达上海的迁移路径,同时,通过对北半球部分国家大气监测数据的总结和分析,对北半球放射性气团的运移路径进行了定量验证。
1 实验部分
1.1 采样地点及条件
气溶胶采样地点设置在上海市普陀区华东师范大学河口海岸大楼楼顶平台(31°13′39″N, 121°23′56″ E),海拔高度约25 m[18]。气溶胶样品的采样信息及气象条件列于表1。
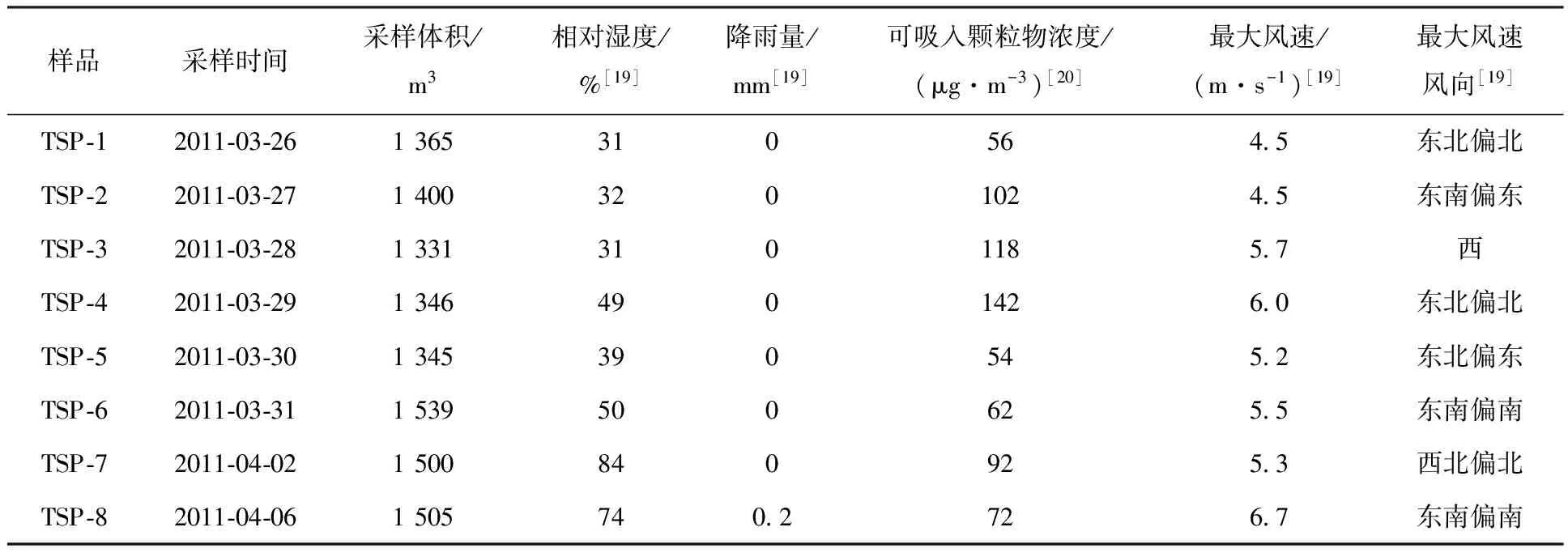
表1 气溶胶样品采样信息及气象条件Table 1 Sampling information of aerosol samples and meteorological conditions
1.2 实验材料与仪器
气溶胶采样仪器是KC-1000型大气TSP采样器(青岛仪器总厂),采集气溶胶所用的滤膜是孔径为0.4 μm聚碳酸酯核孔膜(PET膜,北京清能创新科技有限公司),有效采样面积为18 cm×22.6 cm。
131I和134, 137Cs的γ能谱分析在Canberra BE 3830型HPGe γ能谱仪(Canberra公司)上进行。该仪器的相对探测效率大于35%,系统本底值为0.9 s-1[21]。131I和134,137Cs三种核素对应的能量峰和分支比分别为364.5 keV(81.7%)、604.7 keV(97.6%)和661.6 keV(85.1%)。效率刻度使用无源效率刻度软件(LabSOCS)[22]。
1.3 实验方法及数据处理
采样前滤膜先用pH约为2的盐酸溶液浸泡12 h,然后用超纯水洗至中性,在45 ℃条件下烘干备用[23]。采样过程平均流量控制在1.0 m3/min,时间为24 h,该采样器的捕集效率主要取决于采样的滤膜,本工作中该值大于97%[24-25],采样结束后,将滤膜同样在45 ℃的烘箱中烘干,之后再将膜对折两次(收集面朝内)放入样品盒中,用Canberra BE 3830高纯锗γ能谱仪上机测试。
核素活度均校正到采样日期,除表1中8个TSP样品外,其余气溶胶数据来自于国家核安全局[26]。实验中131I和134, 137Cs三种核素的最低检测限(LLD, mBq/m3)采用以下公式[8]进行计算:
(1)
其中:B表示HPGe γ能谱仪的背景计数(本工作中为131I: 47,134Cs: 10,137Cs: 173);η表示该仪器的探测效率(本工作中为131I: 6%,134Cs: 4%,137Cs: 4%);b表示对应核素的分支比(本工作中为131I: 81.7%,134Cs: 97.6%,137Cs: 85.1%);V表示收集的气溶胶体积,m3(本工作中为1 400 m3);t表示样品的测试时间,s(本工作中为16 895 s)。由公式(1)得,131I和134, 137Cs三种核素的LLD分别为0.19、0.05、0.04 mBq/m3。
2 结果与讨论
实心图形和实线描述数据来自国家核安全局(NNSA)[26],空心图形和虚线描述数据来自实验室监测数据;降雨量数据来自中国气象科学数据共享服务网[19];131I和134,137Cs三种核素实验数据的放射性相对误差范围分别为3.8%~14%,16%~41%和12%~55%□——131I,○——134Cs,△——137Cs,■——131I,●——134Cs,▲——137Cs,◇——131I/137Cs,☆——134Cs/137Cs,◆——131I/137Cs,★——134Cs/137Cs图1 2011年3—4月上海地区大气中131I和134, 137Cs活度浓度(a)、131I/137Cs活度浓度比值和134Cs/137Cs活度浓度比值(b)及降雨量(c)Fig.1 Activities of 131I and 134,137Cs(a), activity ratios of 131I/137Cs and 134Cs/137Cs(b) in the atmosphere and rainfall(c) in Shanghai district in March and April of 2011
2.1 上海地区大气中典型放射性核素、核素浓度比值及降雨量分布2011年福岛核事故期间上海地区的大气气溶胶中131I和134, 137Cs放射性活度浓度随时间变化示于图1。从图1中可以清晰地看出,上海地区2011年3月26日至4月22日期间131I的活度浓度是134, 137Cs活度浓度的2~10倍,且波动性较大,而134Cs和137Cs的活度浓度变化趋势基本一致且比较平缓。本实验在2011年3月27日已检测到大气中较高活度浓度的131I(0.5 mBq/m3),而134Cs和137Cs分别在4月6日和4月8日才被明显检出。对比于上海地区的大气环境本底值(131I: ≤ 0.16 mBq/m3,137Cs: ≤ 0.04 mBq/m3;本实验室2008年4月份数据),131I和134, 137Cs三种核素的活度浓度在监测期间显著升高,这证实福岛核事故已经对上海的大气环境造成了影响,而对于三种核素第一个活度浓度峰值出现的时间,131I(2011-03-28)也明显早于134, 137Cs(2011-04-08)。以上现象一方面与核事故中不同放射性核素的释放量有关,文献[27]报道的福岛核事故气态131I和137Cs释放量分别为130 PBq和12 PBq。另一方面也与放射性核素在输运过程中的传输效应有关:131I的半衰期远远短于两种铯同位素,在传输过程中会大量衰减,加上释放源的不断释放会使大气中的131I发生叠合,因此气溶胶中131I的活度浓度波动比较剧烈,131I/137Cs活度浓度比值的波动也比较大;同时,由于131I的相对原子质量小于两种铯的同位素,且其挥发性强,一般以气态形式与气溶胶紧密结合,致使131I传播速率较快,而两种铯同位素因其具有较大的相对原子质量和较强的颗粒活性,与气溶胶的结合程度不及碘,致使其在随气溶胶传播过程中损失较多,这也造成了福岛核事故后世界很多国家首先检测到的核素一般都是131I的现象[15,28];另外,137Cs和134Cs的物化性质一致,在气溶胶传播时间段内(几天至十几天的时间尺度),两者的半衰期差异(约28 a)可以被忽略,因此,134Cs/137Cs活度浓度比值的变化相对平稳。
131I和134, 137Cs活度浓度的最大值出现在同一天(2011-04-08),分别为1.2、0.6、0.7 mBq/m3,131I活度浓度在2011年3月28日、4月5日、4月10日和4月13日也出现了较为明显的峰值,分别为0.8、0.8、1.0、0.6 mBq/m3,131I的多峰值现象可能是多种来源途径的叠加造成的,同时也从侧面定性反映出核事故中放射性物质的多次释放过程,据文献[27,29]报道,2011年3月12日至4月5日期间持续有放射性物质由反应堆释放进入大气环境,例如2011年3月15日、3月23日和3月30日是报道的三个131I排放峰值日期,对应上文上海地区气溶胶中131I的活度浓度峰值日期(2011-03-28、2011-04-05、2011-04-08、2011-04-10和2011-04-13),时间间隔为11~14 d,说明气溶胶中131I从福岛核电站到达上海地区的时间也在11~14 d的范围。
大气中的131I在传输过程中,快速衰变成为主要控制因素,使得131I/137Cs活度浓度比值随时间变化迅速减小,上海地区从最大比值的10.6至无131I检出(< 0.01 mBq/m3)共经历19 d,相对于福岛核电站附近的筑波市,从最大比值的30.8至无131I检出(<0.01 mBq/m3)的时间超过4个月[30],其变化快但变化幅度小;然而,上海地区两种放射性铯134Cs/137Cs活度浓度比值在1.1附近波动,相对于福岛核电站附近的监测数据1.0[31],其变化幅度不大。
上海地区大气中放射性核素活度还会受到不同气象条件的影响,如表1所示(只列举了本实验室气溶胶采样区间部分),2011年3月份盛行偏北风和偏东风,4月份盛行偏东风[19],即福岛核事故期间,上海地区的盛行风向均有利于大气中的放射性物质由福岛核电站向上海地区的输送;同时,在实验过程和数据分析中发现,当大气中可吸入颗粒物浓度[20]偏低时,检测到的131I和134,137Cs三种核素的活度值也相应偏低,说明二者之间存在一定的相关性;另外,降雨现象基于沉降清除机制[32],会使大气中悬浮的气溶胶和悬浮颗粒物等被大量清除。通过对比中国有关省份在2011年3—4月每日的降雨量分布状况[19],发现上海的地区性降雨成为大气中放射性核素清除的关键性因素。结合图1中降雨量分布图,可以看出2011年4月6日、4月7日和4月22日的降雨冲刷作用在一定程度上会造成大气中放射性核素的沉降。
2.2 放射性物质经大气环流到达上海的输运途径分析
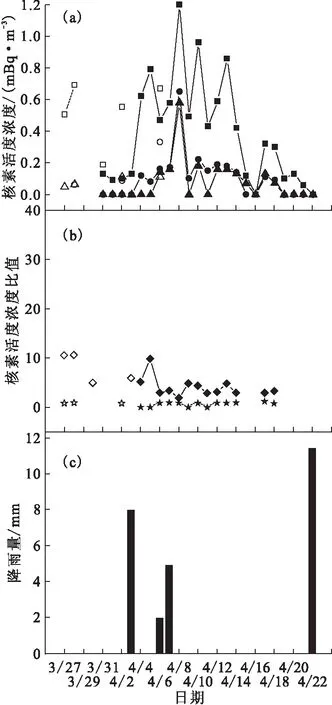
2.2.1 HYSPLIT模型 HYSPLIT模型模拟的后向轨迹(backward trajectory)是指沿时间轴反方向向后返回一段时间的轨迹图,通常被应用于对某一事件发生的原因和过程进行分析。同时,值得说明的是,运移轨迹是由一个起点出发,代表来自该点上空的大气运移途径,途经其他地点后,运移轨迹会发生改变[14]。利用气团运移轨迹模型可以获得气溶胶的输运趋势,但对于放射性气溶胶的输运过程,是气溶胶所携带核素的具体性质以及气象条件等多种因素共同作用的结果。通过HYSPLIT模型模拟的315 h(13 d)后向气团运移轨迹图[33],可以指示2011年3月11日至5月6日期间福岛核事故产生的放射性物质经大气环流到达上海的迁移路径。此处选取2011年3月11日和2011年4月22日两天的后向轨迹图作为典型进行讨论(图2)。依据大气颗粒物的搬运高度和放射性物质的比重[15],同时选择100 m、500 m和3 500 m三个海拔高度来讨论来自福岛核电站的放射性气溶胶到达上海地区的几种可能的迁移途径:总体上的输运趋势,从福岛穿过北太平洋到达美国,穿越北美洲,横跨大西洋,到达北欧国家甚至更北面的北极地区,最终到达上海;到达上海的放射性气团,其中一部分是经亚欧大陆从我国新疆地区由西北路径到达上海,另一部分是经北极地区和俄罗斯从我国东北地区由北向路径到达上海。两种气团运移轨迹的叠加在一定程度上致使上海地区放射性核素的监测结果出现多峰值现象;同时,从轨迹图还可以观察到,西北路径主轴外的边缘轨迹仍然有途经我国东北地区的趋势,因此认为放射性气团的东北径在上海地区的大气放射性监测结果中处于主导的控制地位。
2.2.2 放射性物质在中国境内运移到达上海的两条路径 为了进一步验证2.2.1节中提出的放射性物质从福岛核电站到达上海地区的两条迁移路径,图3中详细列举了两条路径经由的相关省份的131I和134,137Cs活度浓度及131I/137Cs活度浓度比值变化。西北路径的沿途省份自西向东包括:新疆、青海、甘肃、陕西、河南、安徽和上海,东北路径的沿途省份自北向南包括:吉林、辽宁、北京、山东、江苏和上海。从图3可以看出,首先,相比于西北路径中出现的单峰,东北路径中出现了双峰甚至多峰,这种多峰现象体现了福岛核事故过程中放射性物质的多次释放过程[34],而西北路径由于受到北半球整体放射性气团西向输送的影响,在自西向东到达上海的过程中,消减了对于核事故释放过程的表征;其次,西北路径的峰值出现在2011年4月6日、4月7日和4月8日,东北路径的第一波峰值出现在2011年4月3日、4月4日和4月6日,即放射性气团在东北路径上的运移速率更快。基于以上两点,认为上海地区受沿东北路径来的放射性气团的影响程度更大。
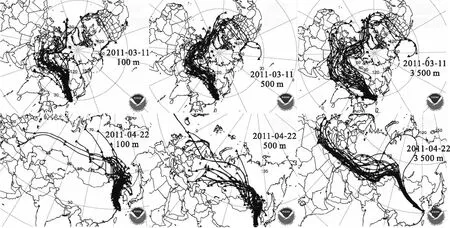
图2 上海(2011-03-11和2011-04-22)315 h后向轨迹分布图[33]Fig.2 Distribution diagram of 315 h backward-trajectories in Shanghai from March 11th to April 22nd, 2011[33]
图3 西北路径(左)和东北路径(右)的131I和134, 137Cs活度浓度及131I/137Cs、134Cs/137Cs活度浓度比值变化[26]Fig.3 Activities of 131I and 134, 137Cs, activity ratios of 131I/137Cs, 134Cs/137Cs in northwest path(left) and northeast path(right)[26]
2.3 北半球大气放射性监测结果
在研究国内大气中放射性物质迁移路径的同时,结合北半球一些国家的报道数据,对北半球大气中131I/137Cs和134Cs/137Cs活度浓度比值的最大值、最大值对应的核素活度浓度值以及最大值出现的日期进行了总结,监测站位示于图4,相关核素活度浓度及核素活度浓度比值归纳于表2。
对于131I/137Cs活度浓度比值,由于131I的挥发性强,与气溶胶等的结合更充分,而颗粒活性较高的137Cs则更倾向于吸附在大气中的颗粒物上,造成大气中131I比137Cs的传输速率快,因此在北半球的监测数据中,同一时间131I比137Cs的活度浓度高出几十甚至几百倍。另外,因为131I的半衰期远小于134, 137Cs,所以在北半球会出现131I活度浓度迅速减小的情况,基于这一点,131I/137Cs活度浓度比值常被用来研究放射性气团迁移过程的时间序列滞后效应问题[28-29,36]。图4和表2中自西向东(除日本外),131I/137Cs活度浓度比值最大值呈减小趋势,对应的时间也呈后向延迟,例如放射性气团到达西雅图(美国)的时间在2011年3月28日之前,即小于事故发生后的17 d,到达维瓦尔城(西班牙)的时间在2011年4月4日之前,而到达新疆(中国)的时间在2011年4月6日之前,即美国到西班牙的时间差是7 d左右,西班牙到中国的时间差是2 d左右,这些结论与前人的研究结果相符[15,35,37]。但是分析中也出现了一些特例,例如相似经度位置的新奥尔松(北极)和维瓦尔城(西班牙),北极最大值出现的时间是2011年3月16日,远早于西班牙,同时北极最大值是西班牙的8~9倍;另外,克拉斯诺雅茨克(俄罗斯)的位置较雅典(希腊)偏东,且两地131I/137Cs活度浓度比值相似,但是俄罗斯出现最大值的时间(2011-04-03)比希腊(2011-04-08)早5 d。这些特例都一定程度上印证了2.2.1节中放射性气团经由北极、俄罗斯,从中国北部抵达上海地区的迁移路径。
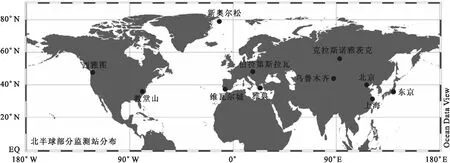
图4 2011年福岛核事故后北半球部分大气监测站分布图Fig.4 Partial air monitoring stations in the northern hemisphere after the Fukushima nuclear accident in 2011
134Cs/137Cs活度浓度比值常被用来区分137Cs的来源,福岛核电站四个反应堆的核燃料成分相同,134Cs产生于反应堆内中子活化反应,所以可以利用二者比值来划分新137Cs和老137Cs[37]。理论上134Cs/137Cs活度浓度比值在放射性物质的运移过程中保持恒定,文献[38]报道的日本福岛核事故期间134Cs/137Cs活度浓度比值在1.00左右,表2中所示大部分国家和地区134Cs/137Cs活度浓度比值大部分也在1.00附近波动。对于二者比值明显偏离1.0的地区,例如教堂山(0.1)和克拉斯诺雅茨克(0.6)等,比值远小于1.0,可以归因于137Cs的历史遗留量的影响,而比值远大于1.0的地区,可能受多种因素影响,其具体原因有待进一步的分析。
3 结 论

(2) 通过HYSPIT气团运移轨迹模型可以定性归纳出放射性气团从福岛核电站到达上海地区的两条迁移路径:西北路径和东北路径。结合我国部分省份公布的大气放射性监测数据,进一步验证了中国境内两条输运途径的存在,同时肯定了东北路径的主要控制地位。地区性大气中放射性核素及其比值的分布规律受到两种来源途径叠加的影响,气象条件、特别是区域性降雨,其清除机制在上海地区也是不可忽略的重要因素。
(3) 北半球大气放射性监测结果中131I/137Cs活度浓度比值的最大值分布呈现自西向东、自北向南的递减趋势,一方面验证了北半球放射性物质自西向东迁移过程中时间序列的滞后效应,另一方面也肯定了放射性物质从北极地区穿过俄罗斯直接到达中国东北部的迁移路径;134Cs/137Cs活度浓度比值在北半球放射性物质迁移过程中保持在相对恒定的1.0,反应了核事故反应堆本身的燃料性质,而对于北半球某些严重偏离该比值的地区,其原因除137Cs的历史遗留因素外,具体原因还需进一步验证。
[1] Amano H, Akiyama M, Bi C, et al. Radiation measurements in the Chiba metropolitan area and radiological aspects of fallout from the Fukushima Dai-ichi Nuclear Power Plants accident[J]. J Environ Radioact, 2012, 111(5): 42-52.
[2] Zheng J, Aono T, Uchida S, et al. Distribution of Pu isotopes in marine sediments in the Pacific 30 km off Fukushima after the Fukushima Daiichi Nuclear Power Plant accident[J]. Geochem J, 2012, 46(4): 361-369.
[3] Baba M. Fukushima accident: what happened?[J]. Radiat Meas, 2013, 55 (709): 17-21.
[4] Koo Y H, Yang Y S, Song K W. Radioactivity release from the Fukushima accident and its consequences: a review[J]. Progress in Nuclear Energy, 2014, 74 (3): 61-70.
[5] Tsumune D, Tsubono T, Aoyama M, et al. Distribution of oceanic137Cs from the Fukushima Dai-ichi Nuclear Power Plant simulated numerically by a regional ocean model[J]. J Environ Radioact, 2012, 111: 100-108.
[6] Povinec P P, Gera M, Holy K, et al. Dispersion of Fukushima radionuclides in the global atmosphere and the ocean[J]. Appl Radiat Isot, 2013, 81(2): 383-392.
[7] Saenko V, Ivanoc V, Tsyb A, et al. The Chernobyl accidente and its consequences[J]. Journal of Clinical Oncology, 2011, 23(4): 234-243.
[8] Wang J, Jiang Y, Huang D, et al. Promotion of the lower limit of detection of gamma emitting nuclides in radioaerosol samples after Fukushima accident[J]. J Radioanal Nucl Chem, 2012, 292(3): 1297-1301.
[9] 王文彩.沙尘气溶胶的传输和气候效应的观测研究[D].兰州:兰州大学,2013.
[11]Menut L, Masson O, Bessagnet B. Contribution of Saharan dust on radionuclide aerosol activity levels in Europe? The 21-22 February 2004 case study[J]. Journal of Geophysical Research, 2009, 114(D16): 1063-1079.
[12]万国江,郑向东,Lee H N,等.黔中气溶胶传输的210Pb和7Be示踪I:周时间尺度的解释[J].地球科学进展,2010,25(5):492-503.
[13]樊元庆,王世联,李慧娟,等.北京地区大气中7Be、137Cs和131I活度浓度分布规律初步研究[J].原子能科学技术,2013,47(2):189-192.
[14]岑况,陈媛,刘舒波,等.日本福岛第一核电站核泄漏后放射性物质运动轨迹[J].地学前缘,2012,19(2):234-238.
[15]Leon J D, Jaffe D A, Kaspar J, et al. Arrival time and magnitude of airborne fission products from the Fukushima, Japan, reactor incident as measured in Seattle, WA, USA[J]. J Environ Radioact, 2011, 102(11): 1032-1038.
[16]Takemura T, Nakamura H, Takagawa M, et al. A numerical simulation of global transport of atmospheric particles emitted from the Fukushima Daiichi Nuclear Power Plant[J]. Sola, 2011, 7(655): 101-104.
[17]Rxler R, Hess G D. An overview of HYSPLIT-4 modeling system for trajectories, dispersion and deposition[J]. Australian Meteorological Magazine, 1998, 47(4): 295-308.
[18]Du J, Zhang J, Zhang J, et al. Deposition patterns of atmospheric7Be and210Pb in coast of East China Sea, Shanghai, China[J]. Atmos Environ, 2008, 42(20): 5101-5109.
[19]数据和产品,地面资料,中国气象科学数据共享服务网,中国科技资源共享网,http:∥www.escience.gov.cn/metdata/page/index.html,2011/3-2011/5.
[20]全国空气质量预报,数据中心,中华人民共和国环境保护部,http:∥datacenter.mep.gov.cn/report/air_daily/air_dairy.jsp,2011/3-2011/5.
[21]吴云峰,杜金洲,黄德坤,等.IAEA国际比对样品的γ谱仪分析[J].核化学与放射化学,2009,31(3):157-162.
[22]黄德坤,吴梅桂,杜金洲.长江口238U、232Th入海通量研究[J].中国核科学技术进展报告,2009,1 (4):235-238.
[23]张安余,张国森,冯冲,等.上海气溶胶中可溶态营养盐含量及影响因素[J].中国环境科学,2013,33(8):1345-1353.
[24]马晓,唐泉,刘永辉,等.滤膜对α-放射性气溶胶取样性能研究[J].核电子学与探测技术,2012,32(1):116-119.
[25]陈亮.大气中放射性气溶胶滤膜过滤效率的测定[J].资源节约与环保,2013(10):66-66.
[26]全国主要城市环境辐射水平,国家核安全局(NNSA),中华人民共和国环境保护部,http:∥www.mep.gov.cn/ztbd/rdzl/dzhaq/xwfb/,2011/3-2011/5.
[27]Masamichi C, Hiromasa N, Haruyasu N, et al. Preliminary estimation of release amounts of131I and137Cs accidentally discharged from the Fukushima Daiichi Nuclear Power Plant into the atmosphere[J]. J Nucl Sci Technol, 2011, 48(7): 1129-1134.
[28]盛黎,周斌,孙明华,等.日本福岛核事故对我国辐射环境影响的监测与分析[J].气象,2013,39(11):1490-1499.
[29]Hideyuki K, Takuya K, Akiko F, et al. Preliminary numerical experiment on oceanic dispersion of131I and137Cs discharged into the ocean because of the Fukushima Daiichi Nuclear Power Plant disaster[J]. J Nucl Sci Technol, 2012, 48(11): 1349-1356.
[30]Kanai Y. Monitoring of aerosols in Tsukuba after Fukushima Nuclear Power Plant incident in 2011[J]. J Environ Radioact, 2012, 111(5): 33-37.
[31]García F P, García M A F. Traces offission products in southeast Spain after the Fukushima nuclear accident[J]. J Environ Radioact, 2012, 114(12): 146-151.
[32]Mitev K, Tsibranski R, Avramov V. Measurements of131I,134Cs and137Cs in environmental samples in Bulgaria after the Fukushima accident[C]∥Nuclear Science Symposium and Medical Imaging Conference (NSS/MIC), IEEE, 2011: 18-24.
[33]HYSPLIT Trajectory Model, HYSPLIT, Air Resources Laboratory. http:∥ready.arl.noaa.gov/hypub-bin/trajasrc.pl, 2011/3-2011/5.
[34]Hou X, Povinec P P, Zhang L, et al. Iodine-129 in seawater offshore Fukushima: distribution, inoganic speciation, sources, and budget[J]. Environ Sci Technol, 2013, 47(7): 3091-3098.
[35]MacMullin S, Giovanetti G K, Green M P, et al. Measurement of airbornefission products in Chapel Hill, NC, USA from the Fukushima Dai-ichi reactor accident[J]. J Environ Radioact, 2011, 112(5): 165-170.
[36]Paatero J, Vira J, Siitari-Kauppi M, et al. Airborne fission products in the high arctic after the Fukushima nuclear accident[J]. J Environ Radioact, 2012, 114(12): 41-47.
[37]Lozano R L, Hernández-Ceballos M A, Adame J A, et al. Radioactive impact of Fukushima accident on the Iberian Peninsula: evolution and plume previous pathway[J]. Environment International, 2011, 37(7): 1259-1264.
[38]Povinec P P, Sykora I, Holy K, et al. Aerosol radioactivity record in Bratislava/Slovakia following the Fukushima accidente: a comparison with global fallout and the Chernobyl accident[J]. J Environ Radioact, 2012, 114(12): 81-88.
[39]Kritidis P, Florou H, Eleftheriadis K, et al. Radioactive pollution in Athens, Greece due to the Fukushima nuclear accident[J]. J Environ Radioact, 2012, 114(12): 100-104.
[40]Bolsunovsky A, Dementyev D. Evidence of the radioactive fallout in the center of Asia (Russia) following the Fukushima nuclear accident[J]. J Environ Radioact, 2011, 102(11): 1062-1064.
[41]Thakur P, Ballarda S, Nelson R. Radioactive fallout in the United States due to the Fukushima Nuclear Plant accident[J]. J Environ Monit, 2012, 14(5): 1317-1324.
华东师范大学 河口海岸学国家重点实验室,上海 200062
利用对气溶胶中典型放射性核素(131I和134, 137Cs)的分析,可以评估福岛核事故产生的放射性物质对上海及全球的大气放射性本底水平造成的影响。本工作结合核事故释放过程、核素的天然衰变以及气象条件等因素,获得核事故期间上海的气溶胶中131I和134, 137Cs活度浓度及其比值的分布特征:131I被检出的时间(2011-03-27)早于134Cs(2011-04-06)和137Cs(2011-04-08),131I的活度浓度(0.01~1.20 mBq/m3)比134Cs(0.01~0.58 mBq/m3)和137Cs(0.01~0.65 mBq/m3)大2~10倍,而且在不同的时间段出现相应的多峰值现象;131I/137Cs活度浓度比值(1.3~10.6)在2011年4月5日之后呈递减趋势,但是134Cs/137Cs活度浓度比值(0.8~2.9)则一直在1.1左右波动。利用HYSPLIT模型模拟放射性气团运移轨迹的分析方法,表明在核事故期间输入到上海的放射性气溶胶的途径有东北和西北两条主要迁移路径。同时通过结合国内相关城市核事故期间大气放射性监测数据,证实了东北路径在中国境内的控制地位。另外,通过总结和分析北半球大气监测数据中131I/137Cs和134Cs/137Cs活度浓度比值最大值的分布特征,验证了日本核事故产生的放射性气溶胶在北半球的传输过程。
气溶胶;131I/137Cs活度浓度比值;134Cs/137Cs活度浓度比值;迁移路径
Source and Pathway of131I,134Cs and137Cs in Aerosols of Shanghai After the Fukushima Nuclear Accident
LIU Dan-tong, WANG Jin-long, BI Qian-qian, DU Jin-zhou*
State Key Laboratory of Estuarine and Coastal Research, East China Normal University, Shanghai 200062, China
The typical radionuclides (131I and134, 137Cs) in aerosols could be efficiently used to evaluate the impacts of radioactive materials from the Fukushima nuclear accident on the radioactive background level of the atmosphere in Shanghai and the world. In the present work, combining impact factors, such as the nuclear accident release process, natural radioactive decay and the meteorological conditions, the distribution patterns of131I and134,137Cs activities and their ratios in Shanghai aerosols were obtained. The date when131I (2011-03-27) could be detectable is earlier than134Cs (2011-04-06) and137Cs (2011-04-08). The131I activity (0.01-1.20 mBq/m3) is 2-10 times higher than those of134Cs (0.01-0.58 mBq/m3) and137Cs (0.01-0.65 mBq/m3). Moreover, multiple peak values of131I are found with time series. The activity ratios of131I/137Cs (1.3-10.6) decrease after the date of April 5th, 2011, however, the activity ratios of134Cs/137Cs (0.8-2.9) are constant to be around 1.1. Using the analytical method of HYSPLIT model, the primary air mass migration pathways of radio-aerosols in Shanghai during this accident can be qualitatively inferred as the northeast and northwest paths. Meanwhile, the northeast pathway of radio-aerosols in Shanghai is further confirmed to be dominant by analysis of the reported radio-aerosol data in the concerned cities of China during that time period. In addition, the results of maximum values of131I/137Cs and134Cs/137Cs activity ratios are summarized and analyzed to reveal the spreading pathways of radio-aerosols in the northern hemisphere.
aerosol;131I/137Cs activity ratio;134Cs/137Cs activity ratio; migration pathway
2015-09-17;
2016-02-29;
时间:2016-09-20
河口海岸学国家重点实验室基本科研业务费资助项目(2011KYYW04)
刘丹彤(1990—),女,山东聊城人,硕士研究生,海洋化学专业
*通信联系人:杜金洲(1964—),男,四川南充人,博士,教授,主要从事同位素海洋学研究,E-mail: jzdu@sklec.ecnu.edu.cn
TL732;TL751
A
0253-9950(2017)01-0103-10
10.7538/hhx.2016.YX.2015073
