CL-20/TNT共晶炸药热分解机理的原子模拟
2017-02-28何远航
刘 海,杨 镇, 何远航
(1.中国空气动力研究与发展中心超高速空气动力学研究所,四川 绵阳 621000;2.北京理工大学爆炸科学与技术国家重点实验室,北京100081)
CL-20/TNT共晶炸药热分解机理的原子模拟
刘 海1,2,杨 镇2, 何远航2
(1.中国空气动力研究与发展中心超高速空气动力学研究所,四川 绵阳 621000;2.北京理工大学爆炸科学与技术国家重点实验室,北京100081)
利用反应力场(ReaxFF)分子动力学方法,研究了CL-20/TNT共晶高温热分解的反应动力学过程与温度和密度的关系;分析了势能和物种数量的演化分布及CL-20和TNT热分解反应的衰减动力学和反应动力学参数。产物识别分析表明,CL-20上-NO2键的断裂是共晶热分解的初始反应路径。随着共晶密度的增加,CL-20和TNT分解的反应能垒均升高。TNT的分解过程对CL-20的分解有抑制作用。N2、H2O、CO2为共晶热分解的最终产物,产生速率大小依次为N2>H2O>CO2。
CL-20/TNT共晶;高温热分解;反应动力学;ReaxFF;反应力场;分子动力学
引 言
含能材料已广泛用于武器战斗部、火箭推进剂等,国内外学者围绕其高效毁伤、安全贮存等问题,在冲击/非冲击条件下的化学反应激发-成长-传播、爆轰等方面进行了广泛研究[1]。但很少涉及原子/分子层面上的化学反应动力学过程,这主要是由于极端条件下含能材料的响应过程涉及较短的时间和空间尺度以及复杂的化学反应动力学过程。加州理工学院Goddard小组提出的反应力场(Reactive Force Field,ReaxFF)分子动力学方法[2],在高温高压条件下传统含能材料(RDX[3]、HMX[4-6]、TATB[4]、PETN[7]、TNT[8]、CL-20[9]等)的热分解研究得到广泛的应用,并针对其中涉及的化学反应动力学过程揭示了很多微观细节。
部分传统含能材料在感度和能量输出方面存在矛盾,如精炼的TNT对摩擦和振动均不敏感,但其在毁伤过程中相对较低的能量输出限制了其在军事领域的应用;CL-20具有较高的爆速、爆压(比HMX高8%~10%),且其能量输出结构具有理想的效果,但其摩擦、撞击以及静电火花感度均高于HMX[10]。塑性黏结、浇铸/固化和压装等手段可提高含CL-20混合炸药的性能。共晶作为一种改性技术,主要通过分子间非共价键将不同种类的含能材料分子结合在同一晶格中[11]。目前,含能材料的共晶尚处于实验室合成阶段,但取得了一定的成果(CL-20/TNT[12]、CL-20/BTF[13]、CL-20/HMX[14]、HMX/TATB[15]、TNT/C10H8[16]),且研究表明共晶含能材料在能量输出以及稳定性方面均有较好的表现。
共晶含能材料爆轰过程的化学反应动力学机制研究是其毁伤效果评估、安全贮存的前提。本研究通过对两种密度的CL-20/TNT共晶炸药施加近似爆轰C-J点温度,并通过反应力场(ReaxFF)分子动力学方法对共晶高温热分解机理进行模拟分析,以期为共晶含能材料爆轰反应过程的理解、新型共晶含能材料合成以及优化共晶内分子配比提供参考。
1 模拟方法及细节
本研究所采用的由CL-20和TNT摩尔比1∶1构成的 CL-20/TNT共晶单胞来自X-射线衍射测定结果[12]。初始单胞内共含有16个分子(8个CL-20分子,8个TNT分子),随后构建3×1×1正交的CL-20/TNT共晶超晶胞结构,共含有48个分子(24个CL-20分子,24个TNT分子),总计1368个原子,密度为1.910g/cm3。为了近似模拟共晶爆轰C-J点的高温高压状态,并分析该点处的化学反应过程[2,4,17],以及初始密度对共晶热分解化学反应动力学过程的影响,通过晶胞体积放大和压缩获得两种密度(1.435g/cm3和2.621g/cm3)的初始共晶结构。两种密度的设置用于比较初始密度对共晶热分解动力学过程的影响。模拟步骤为:首先对初始超晶胞进行结构优化,获得最低总能量结构下的原子位置,随后采用NVT系综弛豫系统内部压力并获得常温下(300K)的晶胞结构,此过程采用Berendsen thermostat方法进行控温,时间步长为0.1fs,此阶段共持续5ps。最后对两种密度下的共晶超晶胞分别施加2000、2500和3000K高温,此过程仍采用NVT系综进行温度控制,各方向均为周期边界条件,计算的时间步长为0.1fs,高温热分解过程共持续50ps。整个过程使用集成ReaxFF/lg势函数[18]程序包的LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator )[19-20]软件进行模拟。图1为CL-20/TNT单胞(a)及3×1×1超晶胞结构(b)。

图1 CL-20/TNT单胞及3×1×1超晶胞结构Fig.1 Crystal structures of unit cell and 3×1×1supercell of CL-20/TNT cocrystal
基于第一性原理的ReaxFF反应力场可以通过键的断裂和形成以及原子间的电荷转移准确地描述大尺度反应系统[2]。Brenner势[21]可以描述键的断裂,但是该势函数不包含范德华和库伦作用。目前,ReaxFF反应力场分子动力学方法已经在含能材料高温热分解[3-5,8]、冲击起爆[6,7,22-26],推进剂燃烧[27-31]等领域得到成功应用,并取得丰富的成果。EReaxFF/lg是在EReaxFF的基础上增加了长程修正项Elg[18],利用EReaxFF/lg势函数对含能材料晶体结构和状态方程的模拟结果与实验结果具有较好的一致性。
2 结果与讨论
2.1 势能和物种数量的演化分布
随着系统温度快速增加至目标温度,反应物首先发生吸热反应而使得系统势能急剧增加,在系统势能达到峰值前,此阶段对应平衡和诱导期,晶胞内发生初级化学反应,具体为—NO2键断裂,NO2不断累积,随后伴随着放热化学反应的进行,系统势能开始衰减,此阶段对应次级反应过程,如图2(a)所示。
初始共晶晶胞内含2种分子,随着热分解的进行,系统内的分子种类随着化学反应的进行而逐渐增多。图2(b)为热分解过程中,晶胞内物种数量随时间的分布曲线。整体来说,在反应前期,随着CL-20和TNT的分解形成各种小分子而使得系统内的物种数量快速增加,之后,伴随着化学反应逐渐趋于平衡而使得系统内物种数量维持在一个动态的平衡阶段。与温度相比,密度对物种数量的影响较大,这主要是由于密度增加,分子(或原子)间距变小,各小分子(或原子)更容易聚合形成大分子或团簇使得系统内的物种类别减少。

图2 共晶热分解势能演化曲线Fig.2 Evolution curves of potential energy for thermal decomposition of cocrystal

图3 共晶热分解物种数量演化曲线Fig.3 Evolution curves of total species for thermal decomposition of cocrystal
2.2 CL-20和TNT的衰减动力学过程
图4为共晶中CL-20和TNT分解演化曲线。

图4 CL-20和TNT分子分解的演化分布Fig.4 Evolution curves of CL-20 and TNT molecules decomposition
由图4可见,当共晶密度为1.435g/cm3时,各温度条件下,TNT完全分解所用时间(2000K∶大于50ps,2500K∶18ps,3000K∶10ps)均大于CL-20分解所用的时间(2000K∶2ps,2500K∶1.5ps,3000K∶1ps),并且随着温度的升高,TNT完全分解的时间明显提前,此结果表明,温度升高会加快共晶中CL-20和TNT的分解,且温度对TNT分解速率的影响大于CL-20。
图5为共晶热分解初级产物NO2随时间的分布演化情况。由图5可见,温度增加会加快共晶热分解的速率,具体表现为:随着温度的升高,NO2达到峰值的时刻提前,并且达到峰值后的衰减速率也明显加快,衰减的原因主要是由于初级产物NO2加入到次级反应中形成N2、H2O和CO2等最终产物。对比图5两种密度条件下NO2的分布特征,密度对NO2的产量具有较明显的影响。高密度情况下,初级反应形成NO2的数量整体上小于低密度的情况,这是由于密度增加,分子间距变小,初级反应形成的NO2会快速加入到次级反应中形成最终产物而使得NO2累积的量减小,并且通过图4比较密度对共晶中CL-20和TNT分解速率的影响,可以推测初级反应形成的NO2来自于共晶中的CL-20。另外,NO2的演化分布与系统势能曲线具有相似的分布特征,因此-NO2键的断裂决定着系统的能量势垒。

图5 初级产物NO2随时间的演化曲线Fig.5 Evolution curves of primary product NO2 with time
共晶中CL-20和TNT分子数量的衰减满足指数函数关系:
C(t)=C0exp(-kt)
(1)
式中:C0为共晶中CL-20和TNT分子的初始数量(24);C(t)为t时刻CL-20或TNT分子的数量;k为衰减速率(1012s-1)。
通过式(1)分别对共晶中CL-20和TNT分子的分解演化曲线进行拟合(仅显示部分拟合曲线,如图4中黑色线所示)得到衰减速率(k)如表1所示。

表1 共晶中CL-20和TNT的衰减速率常数
采用关联反应速率常数(k)和反应活化能(E)的Arrhenius定律对CL-20和TNT衰减动力学过程进行分析:
(2)
式中:T为温度,K,R为理想气体常数;A为指前因子,s-1。
将表1中得到的衰减速率常数代入Arrhenius方程,并对lnk-1/T关系进行线性拟合,结果如图6和表2所示。
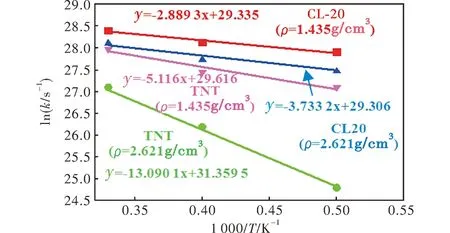
图6 共晶中CL-20和TNT衰减动力学的计算Fig.6 Computation of the decay kinetics of CL-20 and TNT in cocrystal

炸药ρ/(g·cm-3)E/(kJ·mol-1)ln(A/s-1)CL-201.43524.0222.43CL-202.62131.0522.40TNT1.43542.5522.71TNT2.621108.8324.45
在密度相同的情况下,CL-20热分解的反应活化能低于TNT,因此共晶受热后,CL-20分子首先发生分解。此外,随着共晶密度的增加,CL-20和TNT分解的反应能垒均随之升高,并且,密度增加使得TNT分解的反应活化能明显提高。CL-20和TNT分子的衰减速率均随着温度的升高而加快。在相同温度条件下,CL-20分子的衰减速率大于TNT。此外,由表1可知,CL-20分子分解的衰减速率随着密度的增大而降低,而TNT分子分解的衰减速率随着密度的增大而增加。此结果表明,密度增大,TNT分子的衰减速率明显加快,并且TNT的分解抑制了CL-20的分解。
2.3 主要产物识别分析
在产物识别分析中,当任意两个分子碎片中的任意两个原子构成的原子对的键级大于0.3 时,则认为化学键形成,两碎片视为新生成的分子[2]。产物识别结果显示NO2、NO、N2、H2O、CO2为整个热分解过程的主要产物。其中,NO2为初级反应产物,其分布趋势呈现先增加后减少的特征,并且晶胞初始密度对其数量分布影响较大,而温度则主要影响其形成和衰减的速率。NO具有和NO2相似的分布特征。这主要是由于初级产物NO2部分形成NO,并且不稳定的NO最终消耗形成稳定N2。在最终产物形成的过程中,N2的产生速率明显高于CO2和H2O。另外,N2、CO2和H2O同样为CL-20和TNT单晶高温热分解的最终产物[32,8]。

图7 主要产物分布曲线Fig.7 Distribution curves of main products
CL-20/TNT共晶高温热分解最终产物(N2、H2O及CO2)的演化分布同样满足指数函数特征[4]:
C(t)=C∞[1-exp(-kt)]
(3)
式中:C∞为至模拟结束时各主要产物分子数量的渐进分布值;t为时间,ps;C(t)为t时刻对应的分子数量;k为产物形成的速率,ps-1。
利用上述指数函数对共晶热分解最终产物分布曲线(见图7)进行拟合得到N2、H2O以及CO2的产生速率,如表3所示(仅显示部分拟合曲线,如图7(e)、(f)所示)。
整体来说,各温度条件下主要产物的产生速率大小依次为N2>H2O>CO2,并且对于同一温度,低密度下各主要产物的产率小于高密度的产物,且温度和密度的增加均会加快共晶热分解达到终态的速率。

表3 CL-20/TNT共晶最终产物形成的反应速率常数
3 结 论
(1) 利用ReaxFF分子动力学方法模拟了两种密度CL-20/TNT共晶的高温热分解过程。其热分解过程主要分为两个阶段:初级吸热和次级放热过程。上述两个过程有着各自的化学反应特征。从系统势能和NO2的分布曲线具有相似的特征得出,在吸热阶段,反应物发生初级分解反应。并且,CL-20上-NO2脱落是共晶高温热分解的初始反应路径。在系统势能达到峰值后,放热化学反应开始,系统势能不断降低,并逐渐趋于平衡,此时的化学反应特征为初级产物不断被消耗,N2、H2O、CO2等最终反应产物逐渐形成。
(2) 利用Arrhenius定律对CL-20和TNT衰减动力学过程进行分析,结果显示,共晶受热后,CL-20首先发生分解,并形成NO2。共晶密度增加,CL-20和TNT分解的反应活化能均相应升高。同一温度条件下,共晶中CL-20的衰减速率均大于TNT,低密度共晶中CL-20分解的衰减速率大于高密度共晶中CL-20的分解速率;随着共晶密度的增大,TNT的衰减速率明显加快,并且TNT的分解对CL-20的分解有抑制作用。共晶热分解最终产物的形成速率为N2>H2O>CO2。
[1] Fickett W,Davis W C.Detonation: Theory and Experiment[M].New York: Dover Publications,2000.
[2] van Duin A C T,Dasgupta S,Lorant F,et al.ReaxFF: a reactive force field for hydrocarbons[J].The Journal of Physical Chemistry A,2001,105(41): 9396-9409.
[3] Strachan A,Kober E M,van Duin A C T,et al.Thermal decomposition of RDX from reactive molecular dynamics[J].The Journal of Physical Chemistry,2005,122:054502.
[4] Zhang L Z,Zybin S V,van Duin A C T,et al.Carbon cluster formation during thermal decomposition of octahydro-1,3,5,7-tetranitro- 1,3,5,7-tetrazocine and 1,3,5-triamino-2,4,6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations[J].The Journal of Physical Chemistry A,2009,113(40):10619-10640.
[5] Zhou T T,Huang F L.Effects of defects on thermal decomposition of HMX via ReaxFF molecular dynamics simulations[J].The Journal of Physical Chemistry B,2010,115:278-287.
[6] Wen Y S,Xue X G,Zhou X Q,et al.Twin induced sensitivity enhancement of HMX versus shock: a molecular reactive force field simulation[J].The Journal of Physical Chemistry C,2013,117(46): 24368-24374.
[7] Shan T R,Wixom R R,Mattsson A E,et al.Atomistic simulation of orientation dependence in shock-induced initiation of pentaerythritol tetranitrate[J].The Journal of Physical Chemistry B,2013,117(3): 928-936.
[8] 刘海,董晓,何远航.TNT高温热解及含碳团簇形成的反应分子动力学模拟[J].物理化学学报,2014,30(2):232-240.LIU Hai,DONG Xiao,HE Yuan-hang.Carbon-containing formation during pyrolysis of TNT from reactive molecular dynamics simulations [J].Acta Physico-chimica Sinica,2014,30 (2):232-240.
[9] 张力,陈朗,王晨,等.水分子对α 相CL-20 热分解机理影响的分子动力学研究[J].物理化学学报,2013,29(6): 1145-1153.ZHANG Li,CHEN Lang,WANG Chen,et al.Molecular dynamics study of the effect of H2O on the thermal decomposition of α phase CL-20 [J].Acta Physico-Chimica Sinica,2013,29(6): 1145-1153.
[10] 欧育湘,孟征,刘进全.高能量密度化合物CL-20 应用研究进展[J].化工进展,2008,26(12): 1690-1694.OU Yu-xiang,MENG Zheng,LIU Jin-quan.Review of the development of application technologies of CL-20 [J].Chemical Industry and Engineering Progress,2008,26(12): 1690-1694.
[11] Thomas S J M.Crystal engineering: origins,early adventures and some current trends[J].Cryst Eng Comm,2011,13(13): 4304-4306.
[12] Bolton O,Matzger J A.Improved stability and smart-material functionality realized in an energetic cocrystal[J].Angewandte Chemie International Edition,2011,50(38):8960-8963.
[13] Yang Z W,Li H Z,Zhou X Q,et al.Characterization and properties of a novel energetic-energetic cocrystal explosive composed of HNIW and BTF[J].Crystal Growth & Design,2012,12(11): 5155-5158.
[14] Bolton O,Simke L R,Pagoria P F,et al.High power explosive with good sensitivity: A 2∶1 cocrystal of CL-20∶ HMX[J].Crystal Growth & Design,2012,12(9): 4311-4314.
[15] Wei C X,Huang H,Duan X H,et al.Structures and properties prediction of HMX/TATB Co-crystal[J].Propellants,Explosives,Pyrotechnics,2011,36(5): 416-423.
[16] Landenberger K B,Matzger A J.Matzger.Cocrystal engineering of a prototype energetic material: supramolecular chemistry of 2,4,6-trinitrotoluene[J].Crystal Growth & Design,2010,10(12): 5341-5347.
[17] Furman D,Kosloff R,Dubnikova F,et al.Decomposition of condensed phase energetic materials: interplaybetween uni-and bimolecular mechanisms[J].Journal of the American Chemical Society,2014,136:4192-4200.
[18] Liu L C,Liu Y,Zybin S V,et al.ReaxFF-lg: Correction of the ReaxFF reactive force field for london dispersion,with applications to the equations of state for energetic materials [J].The Journal of Physical Chemistry A,2011,115 (40):11016-11022.
[19] Plimpton S.Fast parallel algorithms for short-range molecular dynamics[J].Journal of Computational Physics,1995,117(1): 1-19.
[20] Aktulga H M,Fogarty J C,Pandit S A,et al.Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques[J].Parallel Computing,2012,38(4): 245-259.
[21] Brenner D W.Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films[J].Physical Review B,1990,42(15): 9458.
[22] Nomura K I,Kalia R K,Nakano A,et al.Dynamic transition in the structure of an energetic crystal during chemical reactions at shock front prior to detonation [J].Physical Review Letters,2007,99(14):148303.
[23] An Q,Zybin S V,Goddard III W A,et al.Elucidation of the dynamics for hot-spot initiation at nonuniform interfaces of highly shocked materials[J].Physical Review B,2011,84(22): 220101.
[24] Zhang L Z,Zybin S V,Van Duin A C T,et al.Goddard III.Modeling high rate impact sensitivity of perfect RDX and HMX crystals by ReaxFF reactive dynamics[J].Journal of Energetic Materials,2010,28: 92-127.
[25] Budzien J,Thompson A P,Zybin S V.Reactive molecular dynamics simulations of shock through a single crystal of pentaerythritol tetranitrate[J].The Journal of Physical Chemistry B,2009,113(40):13142-13151.
[26] An Q,Goddard III W A,Zybin S V,et al.Highly shocked polymer bonded explosives at a nonplanar interface: Hot-spot formation leading to detonation[J].The Journal of Physical Chemistry C,2013,117(50): 26551-26561.
[27] Qian H J,van Duin A C T,Morokuma K,et al.Reactive molecular dynamics simulation of fullerene combustion synthesis: ReaxFF vs DFTB potentials[J].Journal of Chemical Theory and Computation,2011,7 (7): 2040-2048.
[28] Weismiller M R,van Duin A C T,Lee J G,et al.ReaxFF reactive force field development and applications for molecular dynamics simulations of ammonia borane dehydrogenation and combustion[J].The Journal of Physical Chemistry A,2010,114(17):5485-5492.
[29] Agrawalla S,van Duin A C T.Development and application of a ReaxFF reactive force field for hydrogen combustion[J].The Journal of Physical Chemistry A,2011,115 (6): 960-972.
[30] Liu L C,Bai C,Sun H.Mechanism and kinetics for the initial steps of pyrolysis and combustion of 1,6-dicyclopropane-2,4-hexyne from ReaxFF reactive dynamics[J].The Journal of Physical Chemistry A,2011,115 (19): 4941-4950.
[31] Chenoweth K,van Duin A C T,Dasgupta S,et al.Initiation mechanisms and kinetics of pyrolysis and combustion of JP-10 hydrocarbon jet fuel[J].The Journal of Physical Chemistry A,2009,113 (9): 1740-1746.
[32] 张力,陈朗,王晨,等.CL-20初始热分解反应机理的分子动力学计算[J].火炸药学报,2012,35(4): 5-9.ZHANG Li,CHEN Lang,WANG Chen,et al.Mechanism of the initial thermal decomposition of CL-20 via molecular dynamics simulation[J].Chinese Journal of Explosives & Propellants(Huozhayao Xuebao),2012,35(4):5-9.
Atomistic Simulation on Pyrolysis Mechanism of CL-20/TNT Cocrystal Explosive
LIU Hai1,2,YANG Zhen2,HE Yuan-hang2
(1.Hypervelocity Aerodynamic Institute,China Aerodynamics Research and Development Center,MianyangSichuan 621000,China;2.State Key Laboratory of Explosion Science and Technology,Beijing Institute of Technology,Beijing 100081,China)
The relationship of reaction kinetic process with temperatures and densities for pyrolysis of CL-20/TNT co-crystal was studied using reactive force field(ReaxFF) molecular dynamics simulation.The evolution distribution of potential energy and total species,decay kinetics and kinetic parameters for thermal decomposition reaction of CL-20 and TNT were analyzed.Product identification analyses show that the breaking of-NO2bond from CL-20 molecules is the initial reaction pathway for thermal decomposition of the cocrystal.With increasing the cocrystal density,the reaction energy barrier of CL-20 and TNT molecule decomposition increases correspondingly.The decomposition process of TNT has an inhibition action on the decomposition of CL-20.Final products for thermal decomposition of the cocrystal are N2,H2O and CO2.The production rate decreases in the order of N2>H2O>CO2.
CL-20/TNT cocrystal; pyrolysis; reactive kinetics;ReaxFF;reactive force field; molecular dynamics
10.14077/j.issn.1007-7812.2017.01.003
2016-07-02;
2016-12-05
刘海(1985-),男,助理研究员,从事爆炸与冲击动力学研究。E-mail: tristan_l@bit.edu.cn
何远航(1964-),男,教授,从事爆炸与冲击动力学研究。E-mail: heyuanhang@bit.edu.cn
TJ55;TQ564
A
1007-7812(2017)01-0014-07
