模拟干湿交替对水稻土古菌群落结构的影响*
2017-02-23包丽君贾仲君
包丽君贾仲君
(1 中国科学院南京土壤研究所,南京 210008)
(2 中国科学院大学,北京 100049)
模拟干湿交替对水稻土古菌群落结构的影响*
包丽君1,2贾仲君1†
(1 中国科学院南京土壤研究所,南京 210008)
(2 中国科学院大学,北京 100049)
干湿交替是自然界普遍存在的现象,但长期以来由于技术的限制,复杂土壤中微生物对水分变化的响应规律仍不清楚。针对我国江苏常熟湖泊底泥发育的典型水稻土,在室内开展湿润-风干以及风干-湿润各三次循环,每次循环中湿润、风干状态各维持7d,利用微生物核糖体rRNA的通用引物进行PCR扩增,通过高通量测序分析土壤古菌多样性变化,同时结合实时荧光定量PCR技术,在DNA和RNA水平研究古菌数量对干湿交替过程的响应规律。结果表明:水稻土湿润-风干过程中,在DNA水平土壤古菌数量降幅约为149倍~468倍,而在RNA水平降幅最高仅为2.06倍;水稻土风干-湿润过程中,在DNA水平古菌数量增幅在147倍~360倍之间,而在RNA水平增幅最高仅为2.95倍。表明在干湿交替过程中,DNA水平的古菌16S rRNA基因数量变化远高于RNA水平。基于高通量测序多样性的结果表明,在DNA和RNA水平,湿润土壤3次风干、以及风干土壤3次加水湿润7d恢复后,土壤古菌群落结构均发生统计显著性改变。在微生物门、纲、目、科和属的不同分类水平下,水稻土古菌主要包括3、10、13、14、10种不同的类群,在RNA和DNA水平的结果基本一致。干湿交替导致部分古菌类群发生显著变化,其中在微生物分类学目水平发生显著变化的古菌最高达到6种,主要包括产甲烷古菌和氨氧化古菌,如Methanobacteriales、Methanosarcinales、Methanomicrobiales和Nitrososphaerales等。这些研究结果表明,反复的干湿交替并未显著改变水稻土中古菌的主要类群组成,古菌类群的绝对数量和相对丰度发生了一定程度的变化,但这些变化与微生物生理作用的联系仍需进一步研究;风干土壤中古菌RNA序列极可能来自于完整的古菌细胞,暗示了这些古菌细胞能够较好地适应水稻土中水分的剧烈变化,风干状态的土壤在一定程度也可用于土壤古菌群落组成研究。
高通量测序;水稻土;16S rRNA;16S rRNA基因;干湿交替;古菌
20世纪以来,由于全球气候变化,包括温度与降水在内的主要气候特征值发生了变化,全球范围内的降水和干旱模式可能持续发生改变[1]。1958年Birch发现干旱的土壤加水培养后,二氧化碳的排放量显著增加,这一现象随后被学术界称为Birch效应或者干土效应[2]。二氧化碳排放量的改变对全球碳循环以及气候变化有着重要影响。然而,Birch效应的基本原理迄今仍未有明确的结论。传统的观点认为Birch效应是一个物理化学过程,当土壤风干后,不同团聚体孔隙中充满大量的空气,而土壤经历降雨后,雨水会逐渐渗入土壤,将闭蓄在土壤中包括CO2等在内的空气压出,造成CO2排放量在短时间内迅速增加[3]。然而,近年来也有研究表明微生物在其中发挥了更大的作用。一般认为,干旱胁迫下微生物细胞能 够自动调节渗透压,积累并产生高浓度的溶质(例如氨基酸、多元醇等)以防止细胞脱水凋亡[3]。而土壤经加水再湿润后,水分迅速渗入导致细胞膨胀裂解,部分细胞溶质外流,从而被其他微生物利用[4],导致风干土壤加水湿润后,微生物呼吸作用增强,短期内CO2排放量显著增加。同时也有报道认为,风干土壤加水湿润时,滞留在团聚体空隙中的空气,由于大气压力差导致土壤团聚体的膨胀裂解,使新的有机质被暴露,从而被微生物利用,土壤呼吸作用增加[5]。微生物种类多、分布广、代谢强、繁殖快,被认为是土壤生态系统中最活跃的部分,是生态系统碳循环主要驱动者[6],对外界环境干扰的响应十分敏感。同时,微生物呼吸作为土壤呼吸的重要组成部分,可能会强烈影响大气二氧化碳浓度,被认为是陆地生态系统光合固定大气碳的几乎唯一输出途径。因此,外界环境如土壤水分的动态变化,不仅会影响土壤微生物群落结构,也会影响生态系统碳氮循环过程[6-7]。稻麦轮作是我国南方重要的水稻管理方式,水稻生长中期烤田也是一种常见农业耕作制度,由此产生的干湿交替过程极可能对土壤微生物群落结构及其活性产生一定的影响,但长期以来由于技术手段的限制,相关研究报道较少。
20世纪80年代,Woese和Fox[8]根据核糖体rRNA序列的系统发育分析,提出了古菌(Archaea)、细菌(Bacteria)和真核生物(Eukarya)的三域分类系统。然而,长期以来,古菌一直被认为仅存在于极端条件下,如极端高盐、强碱或者强酸环境。1992年,研究者利用16S rRNA引物扩增海水微生物群落的基因组DNA,首次发现了非极端环境中存在与极端环境古菌高度相似的“泉古菌”,随后陆地生态系统中也发现存在大量的泉古菌,改变了古菌仅栖息于极端环境的传统认知。据估算,每克土壤最多可能含有约100 亿个微生物细胞,上百万种不同的微生物物种[9],而古菌数量占所有微生物的比例最高可达12%~38%[10],且古菌在土壤元素转化过程中发挥着重要作用,如氨氧化古菌、产甲烷古菌等。然而,古菌对土壤环境中水分变化的适应策略和响应程度研究报道较少,其环境影响规律及驱动机制仍不清楚[11]。
2007年以来,以高通量测序为代表的分子生态学技术快速发展,为研究土壤古菌对干湿交替的适应规律提供了重要的技术支撑。此外,RNA较DNA水平的灵敏度更高,细胞分泌到环境中的游离RNA降解快,通常以秒计,而DNA相对稳定,游离的DNA也可在土壤中保留较长时间,因此,RNA常被用来评价微生物的活性,而DNA通常被用于微生物多样性的资源评价。此外,土壤中的DNA来自于完整细胞内和游离于细胞外的两部分,在一定程度上,DNA分析并不能准确反映细胞水平的微生物种群数量变化,并且土壤DNA提取可能与环境样品的状态、胞外游离DNA数量等紧密相关,单一的实时荧光定量PCR技术无法准确反映土壤微生物种群数量的变化。新一代高通量测序技术的数据通量大、并且能同时分析上百个不同样品,是研究复杂环境中微生物群落结构和物种组成的重要工具[12]。据此,本研究针对江苏常熟典型水稻土,设置了湿润-风干、风干-湿润干湿循环3次,每次持续7d后提取土壤微生物DNA和RNA,通过实时荧光定量PCR和高通量测序多样性技术,研究水稻土古菌对干湿交替过程的响应规律。
1 材料与方法
1.1 土壤样品概况
水稻土采自江苏常熟湖泊底泥发育的土壤(31°33′N,120°42′E),采样小区的农业耕种方式为冬小麦-夏水稻的水旱轮作,2014年4月采集0~20cm表层的新鲜土壤,研磨过2mm筛并保存。土壤的基本理化性质如下:pH7.3,有机质36.2 g kg-1,全氮1.9g kg-1,硝态氮0.04g kg-1,铵态氮0.007g kg-1,土壤最大持水量为53.6%。
1.2 水稻土的干湿交替过程
土壤的干湿交替培养过程具体如下:湿润(F1)-风干(D1)-湿润(F2)-风干(D2)-湿润(F3)-风干(D3)-湿润(F4),其中湿润-风干过程3次,包括F1-D1、F2-D2和F3-D3,风干-湿润过程3次,包括D1-F2、D2-F3和D3-F4。每个过程大约持续7 d并开展分子生态学分析。主要流程如下:首先,称取210g新鲜稻田土壤于无菌托盘置于通风处晾干,将晾干土壤保持7d,随后,将这些风干土壤加水湿润并恢复到新鲜状态保持7d,然后继续进行风干培养7d,并循环培养。每次湿润或风干作为一个处理,每次处理设置3个重复。每个重复收集大约10g土,其中3g土壤样品,加入RNA later (Ambion)后,-20℃保存用于RNA 提取,其余样品保存于-20℃,用于DNA提取以及后续分析。
1.3 水稻土核酸DNA/RNA的提取
利用FastDNA®Spin Kit for Soil试剂盒(MP Bio)提取土壤总DNA,称取干重约为0.5 g的土样,按照试剂盒内说明书的方法操作,提取土壤微生物的总DNA,并溶解于100 μl无菌水后-20℃保存待用。通过微量紫外分光光度计(NanoDrop®ND-1000)测定DNA 浓度和纯度(OD260/OD280和OD260/OD230),其中每个样品浓度均保证30 ng μl-1,大部分DNA样品OD260/OD280值在1.8~2之间以保证DNA质量。同时,利用1.2%的琼脂糖凝胶电泳分析DNA 的完整性和相对浓度。
根据已有文献报道的方法[13],提取土壤微生物的总RNA,具体如下:(1)将保存于-20℃冰箱中,装有等量干重的0.5g土样并加入了500 μlRNA later的2.0 ml带螺帽口的裂解管置于冰上解冻后,13722 rmin-1离心2 min弃上清液,去除RNA later;(2)在土壤沉淀物中加入0.5 g 玻璃珠(0.5mm∶0.1mm=3∶2,Sigma)和预冷的700 μl TPM buffer(50 mmolL-1Tris-HCl pH5.0,1.7% polyvinylpyrrolidone,20 mmolL-1MgCl2),使用Fast PrepTMFP120 核酸提取仪以6 m s-1的速度裂解土壤微生物细胞35 s,随后将裂解混合物在4℃低温下13722 r min-1离心2 min,上清液转移至2.0 ml的无菌离心管;(3)向裂解管中的土壤沉淀物加入预冷的700 μlPBL buffer(5 mmol L-1pH 5.0 Tris-HCl,5 mmolL-1pH 8.0 EDTA Na2,0.1% SDS,6%水饱和酚),重复步骤(2),连续2次提取土壤核酸以提高DNA/RNA回收效率[14];(4)依次利用500 μl的3 种有机试剂清洗(2)(3)步骤合并后的上清液:水饱和酚(p H 4.5),苯酚∶氯仿∶异物醇(25∶24∶1)(pH 4.5),氯仿∶异物醇(24∶1),充分混匀后4℃低温下13722 r min-1离心2 min 并将上清液转移至新的2.0ml无菌离心管中;(5)加入2倍体积PEGNaCl(30% PEG-6000,1.6 molL-1NaCl)于上清液中并混匀,室温下静置2h 后离心10 min,弃上清液后加入400 μl 70% 乙醇清洗沉淀,并再次13 722 r min-1离心5 min;(6)弃上清液后,离心管置于无菌台吹干沉淀并加入50 μlDNase /RNasefree H2O 溶解;(7)利用Recombinant DNaseⅠ(TaKaRa)去除总核酸中的DNA,随后,通过RNeasyMinElute cleanup Kit(QIAGEN)试剂盒纯化土壤总RNA,并利用微生物16S rRNA基因的通用引物(515F/907R)扩增土壤总RNA,确保总RNA中不存在DNA污染;(8)利用微量紫外分光光度计(NanoDrop®ND-1000)测定RNA 浓度,采用PrimeScript 1st strand cDNA Synthesis Kit(Takara)试剂盒将土壤RNA样品反转录成cDNA,并于-20℃保存待用。
1.4 高通量测序土壤微生物16S rRNA及其基因
针对湿润-风干、风干-湿润处理的7个过程,分别提取土壤总RNA和DNA后开展16S rRNA基因和cDNA的多样性分析。如表1所示,首先利用通用引物515F和907R扩增土壤微生物的16S rRNA基因,尽可能无偏差覆盖土壤中的所有微生物群落,以便后续从中提取古菌序列。用于焦磷酸测序的通用引物前端含有12个碱基的特异Barcode标签,用以区分不同的土壤样品。PCR扩增体系为:25 μl的TaKaRa 2×SYBR®Premix EX TaqTM(dNTP Mixture,Buffer,Mg2+Plus)0.5 μl的引物(20 μmolL-1),加入2.0 μl DNA模板和无菌水至50 μl反应体系,每次PCR反应均设置无菌水的阴性对照。PCR扩增条件为:94℃,5.0 min;30× (94℃,30s;55℃,30s;72℃,45s);72℃,10 min。
获得PCR产物后,利用AgaroseMiniBEST DNA Fragment Purification Kit Ver.2.0试剂盒(Takara)切胶纯化,并将纯化产物溶于30μl DNase-free H2O。通过微量紫外分光光度计(NanoDrop®ND-1000)测定浓度后,采用TruSeq Nano DNA LT Sample Prep Kit试剂盒对样品进行建库后,等摩尔数混合,利用Illumina公司MiSeq测序系统上机分析。对测序所得总序列进行去除嵌合体处理,根据不同标签提取样品序列,并进行后续分析。
本研究中3次湿润-风干、风干-湿润过程中共7个处理土壤样品的DNA和cDNA进行Miseq高通量测序,利用Qiime对原始序列进行分析,去除低质量序列后,分别获得1395262和757425条高质量序列,平均每个样品分别获得66441和36067条序列[15]。同时利用Qiime进行系统发育分析,在97%置信度下将每个样品所有的16S rRNA基因序列进行分类,获得门、纲、目、科和属各水平下的分类单元。以其中一个湿润土壤的一个重复样品为例,N为所有高质量序列总和,划分到某个分类单元的序列数为ni,根据公式Pi=ni/ N,得到各个分类单元的相对丰度开展后续分析。进一步通过R软件分别对样品进行群落结构分析。

表1 PCR扩增引物Table 1 Primers used in this study
1.5 土壤总DNA和cDNA的实时荧光定量PCR
实时荧光定量PCR的方法与已有的报道一致[14]。将土壤的总DNA和cDNA样品,根据NanoDrop测定的结果进行稀释,使最终浓度保证在1~10 ng μl-1,作为定量PCR的模板。定量PCR的标线利用含有古菌和细菌16S rRNA基因的克隆进行制备。首先利用特定引物进行目的基因的扩增,构建克隆文库后,将含有目的基因的克隆在LB营养液中过夜培养,提取质粒纯化并测定质粒的浓度,根据摩尔常数计算出目的基因的拷贝数,随后用DNase-free H2O将质粒连续稀释8个数量级,从而得到各个目的基因的标准曲线。实时荧光定量PCR于CFX96 Optical Real-Time Detection System(Bio-Rad)上完成。定量PCR扩增反应体系为:10μl的SYBR®Premix EX TaqTM(Takara),上、下游引物(10 pmolμl-1)各0.5μl,1.0μl土壤总DNA/cDNA模板,加入8μl灭菌双蒸水至20 μl反应体系。每次试验均采用灭菌双蒸水代替DNA 作为反应模板设置严格的阴性对照。
1.6 统计分析
所有数据采用Origin8.1和SPSS16.0进行处理分析,处理之间的平均值差异采用one-way ANOVA单因素方差分析,p<0.05表示显著差异。非度量多维尺度 (NMDS,Non-metric multidimensional scaling)图谱利用R软件统计绘制。
2 结 果
2.1 水稻土干湿交替过程中古菌数量的变化
利用实时荧光定量PCR技术,分别对水稻土3次湿润-风干以及3次风干-湿润过程中古菌数量进行分析。如图1所示,在风干过程中,古菌数量显著降低;而在湿润过程则显著增加。3次风干过程中,古菌数量在DNA水平降幅149倍至468倍,然而,在RNA水平的下降仅发生在第1次和第3次风干过程,降幅最大仅为2.06倍,而在第2次风干过程,古菌rRNA数量基本不变,在统计上未有显著性差异。类似的,湿润过程中,土壤加水湿润后DNA水平古菌数量显著增加,增幅147倍~360倍,而在RNA水平古菌数量的显著增加仅发生在第2次湿润过程中,增幅2.95倍;第1次和第3次加水湿润后,古菌rRNA数量甚至出现了下降趋势。总体而言,风干和湿润过程中古菌群落的16S rRNA及其基因数量呈相反的趋势,尤其在DNA水平古菌数量的变化远远高于RNA水平。
2.2 水稻土干湿交替过程中古菌群落的变化
如表2所示,3次湿润-风干、以及风干-湿润过程中,在DNA和RNA水平分别获得土壤古菌高质量序列共计38839和17213条。在微生物分类门、纲、目、科和属的分类水平下,零时刻湿润土壤F1在DNA水平含有3、9、13、12、8不同的古菌类群,而在RNA水平则包括了3、7、10、9、7种不同的古菌类群。对古菌16S rRNA基因进行非度量多维尺度NMDS分析表明,在DNA和RNA水平下(图2A,图2B),古菌微生物的群落结构均发生了显著性改变(p<0.05)。这一结果表明,本研究中的新鲜土壤样品进行3次风干及加水湿润培养后不仅影响了土壤古菌16S rRNA基因的数量,也对其群落结构有一定的影响。
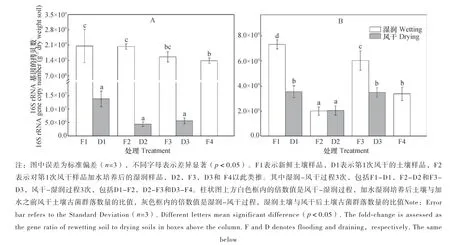
图1 干湿交替下土壤古菌16S rRNA基因在DNA(A)和RNA(B)水平下的数量变化规律Fig. 1 Change of archaeal 16S rRNA genes in abundance at DNA(A)and RNA levels(B),respectively,in soils under wet-dry alternation

表2 MiSeq高通量测序样品描述Table 2 Summary of pyrosequencing results
针对干湿交替过程中显著增加的古菌类群进行分析,如图3所示,风干过程中,在RNA水平下,门、纲、目、科和属水平统计上显著增加的古菌类群明显高于DNA水平。同时,在第3次湿润-风干(FD-3)过程中,在DNA水平未检测到有显著增加的古菌,但是在RNA水平下,门、纲、目、科和属水平均有显著增加的古菌类群。在风干-湿润(DF)过程中,DNA水平下,门、纲、目、科和属水平均有古菌类群发生显著增加,但是在RNA水平却未检测到显著增加的古菌。以第1次湿润-风干过程为例,在DNA水平,在目水平有2个古菌类群发生显著增加,其相对丰度从0.005%,增加了0.023%,增加4.60倍;在RNA水平,目水平有4个古菌类群发生显著增加,其相对丰度从0.786%增加了1.637%,增加2.08倍。
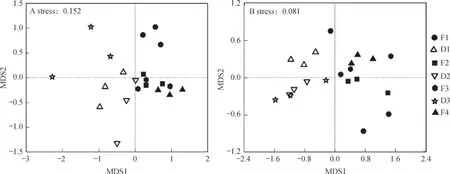
图2 干湿交替过程中古菌微生物16S rRNA基因在DNA(A)和RNA(B)水平下的非度量多维尺度分析Fig. 2 NMDS(non-metric multidimensional scaling)analysis of compositional structure of 16S rRNA genes of the soil archaea under wet-dry alternationat DNA level(A)and RNA level(B),respectively
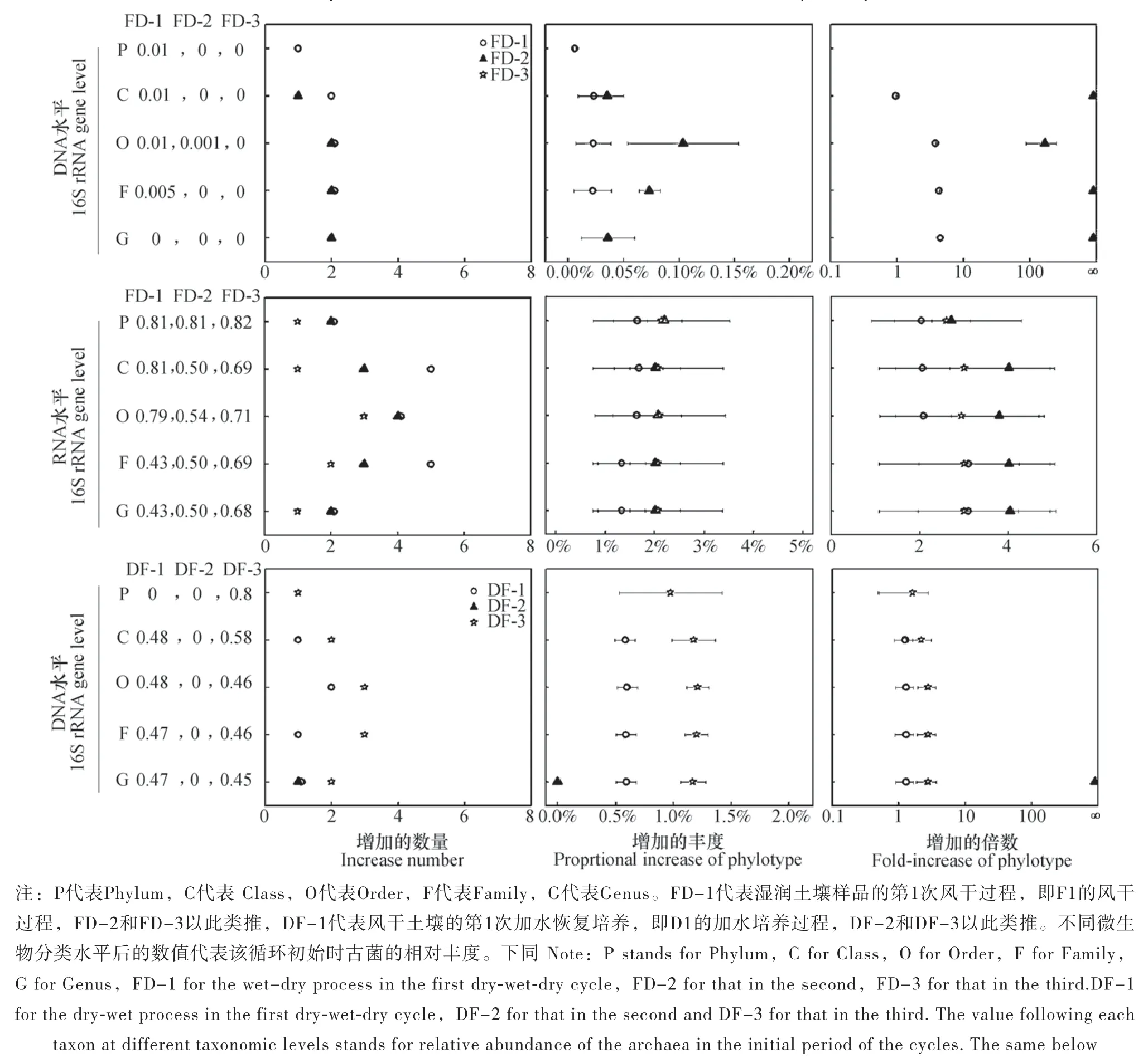
图3 干湿交替过程中显著增加的古菌相对丰度变化规律Fig. 3 Rules of the soil archaea varying in relative abundance with processes of dry-wet alternation
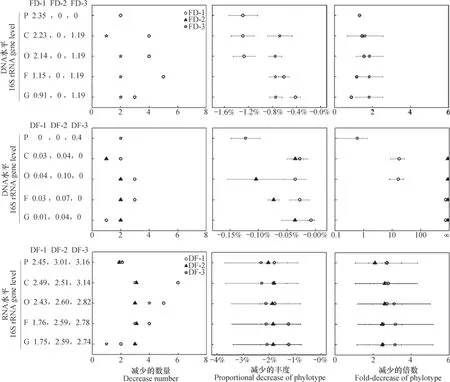
图4 干湿交替过程中显著减少的古菌类群变化规律Fig. 4 Rules of the soil archaea decreasing during the process of wet-dry alternation
进一步针对干湿交替过程中显著降低的古菌类群进行分析,如图4所示,在湿润-风干过程中,在RNA水平未检测到显著减少的古菌,而在DNA水平下,门、纲、目、科和属均检测到显著减少的古菌类群,但DNA水平第2次湿润-风干过程中也未检测到有显著性减少的古菌类群。表明在RNA水平湿润-风干过程中,门、纲、目、科和属水平的不同类群古菌均发生明显增加或未有统计显著变化。同时,在风干-湿润过程中,在DNA和RNA水平下均出现了显著减少的古菌类群,且RNA水平显著减少的古菌数量以及丰度减少值要明显高于DNA水平。结合图3,表明在风干-湿润过程中,在RNA水平下,门、纲、目、科和属水平受显著影响的古菌,相对丰度均是降低。
2.3 水稻土干湿交替过程中古菌主要组成的变化
如图5所示,干湿交替过程中,水稻土古菌主要包括泉古菌门(Crenarchaeota)和广古菌门(Euryarchaeota),而前者以氨氧化古菌为主,后者则主要由产甲烷古菌组成。在微生物分类目的水平,水稻土在D N A和R N A水平分别包括了1 4和1 3种主要功能群,主要包括pGrfC26和Nitrososphaerales类群(泉古菌门),Methanosarcinales、Methanobacteriales、YCE6、E2和Methanocellales(广古菌门),这些功能群占水稻土古菌百分比高达80%以上。此外,在风干处理的DNA样品中,也检测到耐盐古菌Halobacteriales,其相对丰度随着风干次数的增加而不断增长,但在RNA水平未能检测到耐盐古菌。

图5 干湿交替过程中土壤古菌在目水平下分类单元的相对丰度。A和B分别是在DNA和RNA水平下的高通量序列分析Fig. 5 Variation of the soil archaea at order level in relative abundance during the processes of dry-wet alternation,and high-throughput sequencing at DNA level(A)and RNA level(B),respectively
在微生物分类目水平下,产甲烷古菌(Methanosarcinales和Methanobacteriales)和泉古菌(pGrfC26和Nitrososphaerales)对土壤风干和加水湿润的响应模式各有不同。如图6所示,在湿润-风干过程中,Methanobacteriales 在DNA水平丰度有增加趋势,却无显著差异,但在R N A水平的丰度显著增加(图6 A)。Methanosarcinales在DNA水平丰度显著减少,而在RNA水平丰度有增加趋势,却无显著差异(图6B)。Nitrososphaerales和pGrfC26在DNA水平丰度均出现过显著降低,而在RNA水平正好相反,丰度呈现增加趋势(图6C,图6D)。在风干-湿润过程中Methanobacteriales、Methanosarcinales、Nitrososphaerales和pGrfC26在DNA和RNA水平丰度的变化与湿润-风干过程正好相反(图6E—图6H)。以Methanobacteriales为例,在DNA水平,湿润-风干过程中,Methanobacteriales丰度有增加趋势,却无显著差异,而在风干-湿润过程中正好相反,Methanobacteriales丰度有减少趋势,却无显著差异;在RNA水平,湿润-风干过程中,Methanobacteriales丰度显著增加,而在风干-湿润过程中,Methanobacteriales丰度显著降低(图6E)。
3 讨 论
干湿交替是土壤中普遍发生的一种自然现象,它对于全球元素循环有着重要的影响[18-19],且古菌在自然界重要元素的生物地球化学循环过程中发挥着重要作用。故本文就湿润土壤的风干过程以及风干土壤的加水湿润过程对古菌的影响展开了相关研究。在湿润-风干(FD)过程中,古菌数量在DNA水平降幅要高于在RNA水平的降幅;在风干-湿润(DF)过程中,DNA水平古菌数量增幅要明显高于RNA水平的增幅,表明水分变化对古菌表达的影响要小于对其数量的影响。高通量测序结果表明,DNA和RNA水平,H2/CO2利用型的产甲烷古菌Methanobacteriales在湿润-风干过程中,相对丰度有增加趋势,且在RNA水平显著增加;在风干-湿润过程,相对丰度有减少趋势,且在RNA水平显著减少,说明Methanobacteriales在土壤水分匮乏时具有一定的耐受性,而当土壤水分恢复时,具有较强缓冲能力。而乙酸利用型产甲烷古菌Methanosarcinales相对丰度在DNA水平显著减少,在RNA水平无显著差异,表明在水分胁迫下,Methanosarcinales仍然能够正常表达。氨氧化古菌Nitrososphaerales在DNA水平相对丰度显著减少,而在RNA水平显著增加,表明风干过程对Nitrososphaerales数量有一定影响的减少,但并没有响应其表达。在湿润-风干过程,风干土壤中古菌RNA水平明显增加,表明古菌细胞能够保持一定的完整性,并在适应水分胁迫的过程中大量表达。此外,我们在水稻土中检测到了与极端嗜盐古菌Halobacteria高度类似的16S rRNA序列,而极端嗜盐古菌Halobacteria主要分布于天然或人工的高盐环境,如盐(碱)湖、晒盐场、盐矿、人工腌制品等。此外,干湿交替过程中也检测到与海洋古菌MCG(Miscellaneous Crenarchaea Group)高度相似的序列,MCG通常分布在海洋底泥,可能参与全球范围内的沉积作用,同时具有较低的呼吸速率[20]。这些结果在一定程度上表明,古菌在地球环境中的分布极为广泛,并且这些古菌对环境水分的变化具有较强的适应能力。
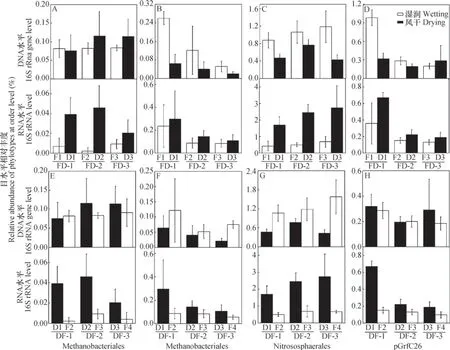
图6 干湿交替过程中古菌主要组成的变化规律Fig. 6 Rules of the soil archaea varying in composition during the wet-dry alternation
实时荧光定量PCR的结果表明,风干过程均会使DNA水平的古菌16S rRNA基因拷贝数显著性降低,而风干土壤加水湿润培养后,土壤古菌16S rRNA基因的拷贝数均又出现了明显的增加,这与已有的研究报道保持一致[21-22]。根据16S rRNA基因拷贝数推测,3次风干土壤加水恢复培养后,DNA水平古菌数量变化要远远高于RNA水平数量变化。干湿交替过程中古菌RNA丰度变化较小,其原因可能与古菌对极端环境如干旱、强酸、强碱和高盐等具有较强的适应能力有关[23]。早期有研究显示,细胞通过植物进行休眠阶段的代谢转换适应干旱[24]。同时,有研究证实,休眠中的细胞仍然有大量核酸的累积,有利于古菌细胞延长生存,快速恢复代谢活性等[25]。这些研究均暗示,土壤风干过程可能导致部分细胞处于休眠状态,其转录丰度变化不明显。值得注意的是,干湿交替过程中古菌16S rRNA基因数量变化高达468倍,一方面可能是由于古菌迅速增长繁殖,但另一方面也可能是DNA提取中存在一定的实验误差。例如,风干土壤加水湿润土壤DNA提取极可能与其状态紧密相关,湿润土壤和风干土壤的DNA提取效率可能有着极大差异,并可能显著影响后续的实时荧光定量PCR结果。此外,游离DNA的存在会高估土壤中古菌的16S rRNA基因数量,而湿润土壤和风干土壤中的游离DNA提取效率可能具有极大的差异,导致干湿交替过程古菌16S rRNA基因数量的显著差异。复杂水稻土中,古菌如何适应水分的剧烈变化过程,在 DNA和RNA水平的响应和适应机制仍然需要进一步研究[26]。
一般认为,产甲烷古菌是严格厌氧微生物,但值得注意的是,本实验在完全好氧条件下进行,产甲烷古菌尽管在数量上可能发生明显变化,但产甲烷生理过程几乎不可能进行。因此,产甲烷古菌数量的增加或者减少,极可能与其生理活性关联度较小。以H2和CO2利用型产甲烷古菌Methanobacteriales为例,湿润-风干过程中,在RNA水平古菌的相对丰度显著增加,而产甲烷古菌失水过程中似乎很难具有生理活性,因此,RNA表达量的增加极有可能来自于产甲烷古菌对水分胁迫的响应。此外,RNA降解极快,风干土壤中几乎不可能以游离状态存在,因此,风干土壤中RNA极有可能来自于完整的古菌细胞。古菌在RNA水平相对丰度的增加,在一定程度上表明风干土壤中产甲烷古菌的细胞能够保持一定的完整性,并在适应水分逐渐丧失的胁迫过程中大量表达。相应的,氨氧化古菌也表现出了类似的规律,在湿润-风干过程中,RNA水平的相对丰度明显增加,表明风干土壤中可能存在较为完整的氨氧化古菌细胞并能够较好地适应水分丧失的胁迫过程。随着单细胞技术的快速发展,结合其他一些先进的物理化学方法,未来将有可能在更精细化的水平准确描述复杂土壤中古菌细胞对干湿交替过程的适应和响应规律。
一般而言,自然环境中土壤微生物群落的形成是一个长期的过程,特定土壤中的微生物群落在组成上也具有相对的稳定性。因此,在短期的干湿交替条件下培养,土壤中不同的微生物物种可能具有不同的生长策略和代谢速率,如在加水恢复培养后个别微生物繁殖较快并能迅速成为优势种群,但短期培养似乎很难改变绝大部分微生物,如古菌的物种组成。我们推测实验土壤中的古菌微生物是其长期适应自然环境变化的演化产物。然而,干湿交替过程中,本研究发现DNA和RNA水平下古菌数量变化规律具有极为显著的差异,相差可能达到上百倍。由此带来的问题是,风干-湿润过程中,古菌数量在DNA水平的增幅高达数百倍,这种极显著的数量变化是否来自于古菌细胞的大量繁殖,抑或是土壤DNA提取效率问题,目前仍不得而知。然而,一般认为DNA片段在土壤中的存留时间较长,甚至可保存几十年,而RNA降解快,因此RNA极有可能来自于完整的细胞,而DNA也可能来自土壤中游离的DNA,风干-湿润的加水湿润过程中促进了土壤DNA提取效率,导致古菌16S rRNA基因的拷贝数增加上百倍。未来仍需开发先进的技术方法,减少土壤中游离DNA的干扰,更加准确地表征土壤微生物多样性及其对干湿交替等环境干扰的响应和适应机制。
4 结 论
新鲜水稻土经过3次干湿交替处理后,古菌数量变化明显,同时在微生物分类门、纲、目、科和属各水平均有不同古菌发生显著改变,进而可能改变土壤微生物群落结构。本研究表明风干以及加水恢复过程对古菌数量和活性均有一定程度的影响,但未来仍需在细胞水平准确刻画复杂土壤环境中古菌对水分变化的适应和响应规律。例如,干湿交替过程中古菌绝对数量的剧烈变化,是否来源于古菌适应水分变化的生理生长,抑或是来自于DNA/ RNA提取等分子技术的误差。此外,干湿交替过程对古菌细胞活性的影响也是未来研究重点之一。
[1]雷 Wen,查尔斯 A Lin. 全球气候变化及其影响. 水科学进展,2003,14(5):667—674 Lei Wen,Charles A L. Global climate change and its impacts(In Chinese). Advances in Water Science,2003,14(5):667—674
[2]Brich H F. The effect of soil drying on humus decomposition and nitrogen availability. Plant and Soil,1958,10(1):9—31
[3]Fierer N,Schimel J P. A proposed mechanism for the pulse in carbon dioxide production commonly observed following the rapid rewetting of a dry soil. Soil Science Society of America Journal,2003,67(3):798—805
[4]Schimel J,Balser T C,Wallenstein M. Microbial stressresponse physiology and its implications for ecosystemfunction. Ecology,2007,88(6):1386—1394
[5]Denef K,Six J,Bossuyt H,et al. Influence of drywet cycles on the interrelationship between aggregate,particulate organic matter,and microbial community dynamics. Soil Biology & Biochemistry,2001,33 (12/13):1599—1611
[6]Balser T C,Firestone M K. Linking microbial community composition and soil processes in a california annual grassland and mixed-conifer forest. Biogeochemistry,2005,73(2):395—415
[7]Sheik C S,Beasley W H,Elshahed M S,et al. Effect of warming and drought on grassland microbial communities. The ISME Journal,2011,5(10):1692—1700
[8]Woese C R,Fox G E. Phylogenetic structure of prokaryotic domain:The primary kingdoms. Proceedings of the National Academy of Sciences of the United States of America,1977,74(11):5088—5090
[9]Gans J,Wolinsky M,Dunbar J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science,2005,309(5739):1387—1390
[10]Kemnitz D,Kolb S,Conrad R. High abundance of Crenarchaeota in a temperate acidic forest soil. FEMS Microbiology Ecology,2007,60(3):442—448
[11]Placella S A,Brodie E L,Firestone M K. Rainfallinduced carbon dioxide pulses result from sequential resuscitation of phylogenetically clustered microbial groups. Proceedings of the National Academy of Sciences of the United States of America,2012,109(27):10931—10936
[12]Rinke C,Schwientek P,Sczyrba A,et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature,2013,499(7459):431—437
[13]Mettel C,Kim Y,Shrestha P M,et al. Extraction of mRNA from soil. Applied and Environmental Microbiology,2010,76(17):5995—6000
[14]郭赟,吴宇澄,林先贵,等. 3次连续重复提取DNA能较好反映土壤微生物丰度. 微生物学报,2012,52 (7):894—901
Guo Y,Wu Y C,Lin X G,et al. Three successive extractions reduce quantification bias of microbial communities associated with incomplete DNA recovery in soil(In Chinese). Acta Microbiologica Sinica,2012,52(7):894—901
[15]Caporaso J G,Kuczynski J,Stombaugh J,et al. Qiime allows analysis of high-throughput community sequencing data. Nature Methods,2010,7(5):335—336
[16]Stubner S. Enumeration of 16S rDNA of Desulfotomaculum lineage 1 in rice field soil by real-time PCR with SybrGreenTMdetection. Journal of Microbiological Methods,2002,50(2):155—164
[17]Francis C A,Roberts K J,Beman J M,et al. Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proceedings of the National Academy of Sciences of the United States of America,2005,102(41):14683—14688
[18]Miller A E,Schimel J P,Meixner T,et al. Episodic rewetting enhances carbon and nitrogen release from chaparral soils. Soil Biology & Biochemistry,2005,37 (12):2195—2204
[19]Warren C R. Response of osmolytes in soil to drying and rewetting. Soil Biology & Biochemistry,2014,70:22—32
[20]Kubo K,Lloyd K G,Biddle J F,et al. Archaea of the Miscellaneous Crenarchaeotal Group are abundant,diverse and widespread in marine sediments. The ISME Journal,2012,6(10):1949—1965
[21]Alvarez H M,Silva R A,Cesari A C,et al. Physiological and morphological responses of the soil bacterium Rhodococcus opacus strain PD630 to water stress. FEMS Microbiology Ecology,2004,50(2):75—86
[22]Potts M. Desiccation tolerance of prokaryotes. Microbiological Reviews,1994,58(4):755—805
[23]Valentine D L. Adaptations to energy stress dictate the ecology and evolution of the archaea. Nature Reviews Microbiology,2007,5(4):316—323
[24]Pnueli L,Hallak-Herr E,Rozenberg M,et al. Molecular and biochemical mechanisms associated with dormancy and drought tolerance in the desert legume Retama raetam. The Plant Journal,2002,31(3):319—330
[25]Sukenik A,Kaplan-Levy R N,Welch J M,et al. Massive multiplication of genome and ribosomes in dormant cells(akinetes)of Aphanizomenon ovalisporum(Cyanobacteria). The ISME Journal,2012,6(3):670—679
[26]van Bodegom P. Microbial maintenance:A critical review on its quantification. Microbial Ecology,2007,53(4):513-523
(责任编辑:卢 萍)
《土壤学报》2015年度优秀论文评选揭晓
《土壤学报》2015年度优秀论文评选活动已于近期结束。经本刊编委推荐和评选,共评出优秀论文奖9篇,涵盖土壤地理与土壤信息、土壤物理与土壤侵蚀、土壤化学、土壤生物、植物营养、土壤肥力、土壤管理等版块。现将获奖名单公布如下(详见附件),并授予年度优秀论文证书,给予适当奖励。
附件:
《土壤学报》编辑部
二〇一六年十二月二日
Changes of Archaeal Communities in a Paddy Soil under Drying and Re-wetting Cycles
BAO Lijun1,2JIA Zhongjun1†
(1 Institute of Soil Science,Chinese Academy of Sciences,Nanjing 210008,China)
(2 University of Chinese Academy of Sciences,Beijing 100049,China)
【Objective】Dry-wet alternation is a common phenomenon existing in nature,especially in paddy soil. But it is still unclear about rule of the response of soil microorganisms to such variation of soil moisture in complex soil,because of limitations in technology. This study was aimed to investigate rules of changes inabundance and community of archaea in a typical paddy soil derived frnom lacustrine sediment,in Changshu of Jiangsu,experiencing three cycles of drying and wetting in lab. 【Method】Soil samples of the typical paddy soil from Changshu were subjected to three consective cycles of drying and wetting in lab. Each of the drying or wetting period lasted 7 days. Real-time quantitative polymerase chain reaction(Realtime PCR)and high-throughput sequencing of 16S rRNA genes were performed to analyze how the archaea in the soil samples changed in biodiversity,abundance and community in response to the dry-wet alternations at DNA and RNA levels. 【Result】The Real-time PCR analysis suggests that the abundance of archaeal 16S rRNA genes copy number changed in response to dry-wet alternation,dropping at DNA level by 149~468 times,but only by 2.06 times on RNA level during the process from wet to dry,and rising by 147~360 times at DNA level,but only by 2.95 times at RNA level during the process from dry to wet. These findings indicate that the change of archaea in 16S rRNA genes copy number was far greater at DNA level than at RNA level. Based on the high-throughput sequencing of 16S rRNA genes,it was found that the archaea community changed significantly in structure at both DNA level and RNA level after three consecutive dry-wet cycles and the changes may be described by non-metric multidimensional scaling(NMDS)(p<0.05). The archaea in the paddy soil could be sorted into 3,10,13,14 and 10 groups at either DNA or RNA level,when classified at phylum,class,order,family and genus level,respectively. The alternation caused significant changes in the archaeal community,especially the six groups at order level,including mainly Methanogenicarchaea and ammonia-oxidizing archaea,like Methanobacteriales,Methanosarcinales,Methanomicrobiales and Nitrososphaerales. The change varied by 2.1%,from 2.82% to 0.69% in total abundance and by 3.79 times,from 0.54% to 2.60%,in maximum fold. 【Conclusion】The Real-time PCR analysis demonstrates that the abundance of archaeal 16S rRNA genes copy number changed significantly in response to the alternation. The high-throughput sequencing of 16S rRNA genes indicates that the archaeal community in the soil changed significantly in structure with the soil alternating in soil moisture condition from dry to wet and from wet to dry. As in environment free extracellular RNAs decompose rapidly,while free extracellular DNAs may remain intact for quite a long time,it is quite probable that archaeal RNA sequence may come from intact microbial cells,and these archaeal cells are able to adapt to severe moisture changes in paddy soil. As the process of drying or wetting does have some impacts on abundance and composition of archaea communities,it is moreadvisable to unfold studies on changes in soil archaea at DNA and RNA levels simulataneously so as to expose rules of the soil archaea responding to dry-wet alternation of the soil.
High-throughput sequencing;Paddy soil;16S rRNA;16S rRNA gene;Dry-wet alternation;Archaea

《土壤学报》2015年度优秀论文奖获奖名单(以版块顺序排序)
Q938
A
10.11766/trxb201603140046
* 国家重点基础研究发展计划(973计划)项目(2015CB150501)资助 Supportedbythe National Key Basic Research Programof China(No.2015CB150501)
† 通讯作者Correspondingauthor,E-mail:jia@issas.ac.cn
包丽君(1990—),女,安徽安庆人,硕士研究生,主要从事微生物生态研究。E-mail:ljbao@issas.ac.cn
2016-03-14;
2016-07-04;优先数字出版日期(www.cnki.net):2016-11-08
