HPLC法测定百合固金颗粒中芍药苷和甘草苷
2017-02-16张鑫王云霞
张鑫,王云霞
(西安市食品药品检验所,西安 710054)
HPLC法测定百合固金颗粒中芍药苷和甘草苷
张鑫,王云霞
(西安市食品药品检验所,西安 710054)
建立同时测定百合固金颗粒中芍药苷和甘草苷含量的高效液相色谱法。采用Welch UltimateXB–C18色谱柱(250 mm×4.6 mm,5 μm),流动相为乙腈–0.1%磷酸(体积比为14∶86),流速为1.0 mL/min,柱温为30℃,检测波长为230 nm。百合固金颗粒中芍药苷在14.91~149.12 ng,甘草苷在34.75~347.52 ng范围内均有良好线性,线性相关系数分别为0.999 5,0.999 2,平均回收率分别为96.7%,95.9%,测定结果的相对标准偏差小于3%(n=6)。该方法操作简单,专属性强,准确度高,重复性好,可以有效地控制百合固金颗粒的质量。
百合固金颗粒;芍药苷;甘草苷;含量测定;HPLC
百合固金颗粒源于处方百合固金汤[1–3],是由百合、地黄、熟地黄、麦冬、玄参、白芍、甘草等十味药加辅料而制成,具有养阴润肺、化痰止咳作用。临床主要用于治疗肺肾阴虚、干咳少痰、咽干吼痛等症状。处方中的百合主要用于润肺止咳,白芍用于柔肝止痛,甘草具有缓急止痛,调和诸药等作用。国家标准中甘草和白芍的处方量是相同的,白芍主要是通过测定芍药苷的含量来控制其药品的质量[4–6],甘草是以薄层色谱进行鉴别[7]。由于处方中中药成分较多,单一成分的含量不能很好地反应药品内在质量,因此笔者提出通过同时测定白芍中的芍药苷和甘草中的甘草苷来有效控制药品质量。
目前对芍药苷和甘草苷的测定主要有高效液相色谱法[8–12]、近红外光谱法[13]、高效液相串联质谱法等[14],尚未有同时测定百合固金颗粒中芍药苷和甘草苷含量的报道。笔者采用HPLC法同时测定百合固金颗粒中芍药苷和甘草苷的含量,比目前标准中通过单一测定来控制药品质量更全面,该方法能够更完善地反映药品中的活性成分,为更好地控制药品质量提供了依据。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪;Agilent1260型,配有四元洗脱泵,全自动进样器,美国安捷伦公司;
紫外可见分光光度计:UV2700型,日本岛津仪器公司;
电子天平:BT124S型,德国赛多利斯公司;
双频数控超声波清洗器:KQ–70VDB型,昆山市超声仪器有限公司;
百合固金颗粒:批号为150905,151002,151106,威海迪沙药业集团有限公司;
芍药苷(批号110736–201136)甘草苷(批号111610–201106):购于中国食品药品鉴定研究院;
乙腈:色谱纯,美国Merck公司;
纯净水:杭州娃哈哈集团有限公司;
实验所用试剂均为分析纯。
1.2 仪器工作条件
色谱柱:Welch UltimateXB–C18柱(250 mm×4.6 mm,5 µm);流动相:乙腈–0.1%磷酸(体积比14∶86);流速:1.0 mL/min;柱温:30℃;检测波长:230 nm;进样量:10 µL。
在上述色谱条件下测定对照品、供试品溶液及阴性样品溶液。理论塔板数以芍药苷峰计算不低于2000,分离度大于1.5,色谱图见图1。

图1 对照品、供试品和阴性样品的液相色谱图
1.3 对照品溶液的制备
精密称取芍药苷7.24 mg、甘草苷4.66 mg分别置于50 mL容量瓶中,加70%乙醇稀释至标线,作为对照品储备液。分别精密吸取芍药苷储备液3 mL、甘草苷储备液2 mL置于25 mL容量瓶中,加如70%乙醇稀释至标线。得对照品溶液,芍药苷的质量浓度为17.376 µg/mL,甘草苷的质量浓度为7.457 µg/mL。
1.4 供试品溶液的制备
取样品10袋,混匀,研细,精密称取1.0 g,置于具塞锥形瓶中。精密加入70%乙醇25 mL,密塞,称量。超声处理30 min,放冷,再称量,用70%乙醇补足减失的质量,摇匀,用0.45 µm的滤膜滤过,取续滤液作为供试品溶液。
1.5 阴性样品溶液的制备
按照处方工艺和处方量,取缺白芍和甘草的药材作为阴性样品,按供试品溶液的制备方法制成阴性对照溶液。分别吸取对照品和供试品溶液及阴性样品溶液各10 µL,进样分析,结果表明所制备的阴性样品溶液对芍药苷和甘草苷的测定无干扰。
2 结果与讨论
2.1 提取方法的选择
试验了不同提取时间(0.5 ,1.0 ,2.0 h)时甲醇和不同浓度的乙醇(30%,50%,70%乙醇)的提取效果,并比较了相同条件下,索氏、回流与超声3种方法的提取效果。结果发现3种方法提取效果差异不大,用70%乙醇提取0.5 h时效果最佳。由于超声提取方法操作简单,故实验采用70%乙醇超声0.5 h对样品进行提取。
2.2 流动相的选择
测定条件的选择主要参考2015版中国药典[15]白芍、甘草含量测定条件,并对甲醇–水、甲醇–磷酸、乙腈–水及乙腈–0.1%磷酸流动相进行了试验考察。结果发现以乙腈–0.1%磷酸为流动相时,供试品溶液色谱图基线平稳,理论塔板数高,峰形较好,分离度高,因此实验选择以乙腈–0.1%磷酸(14∶84)为流动相。
2.3 检测波长的选择
在190~500 nm处,对芍药苷和甘草苷对照品溶液进行扫描,结果发现230 nm波长下两者具有较好的吸收且峰形相对尖锐,干扰较小,故确定检测波长为230 nm。
2.4 标准工作曲线方程及检出限
分别精密吸取1.3中对照品溶液2,4,8,12,20 µL注入液相色谱仪,按照1.2色谱条件进行测定,以待测组分的量(X,ng)为横坐标,相对应的色谱峰面积(Y)为纵坐标进行线性回归,线性方程、相关系数见表1。
精密吸取1.4中供试品溶液,用70%乙醇逐步稀释,按照1.2的色谱条件进样,当S/N=3时,测得芍药苷和甘草苷的检出限分别为2.58,2.23 µg/g,见表1。

表1 线性方程、相关系数及检出限
2.5 精密度试验
移取1.4中供试品溶液,连续进样6次,进行测定。结果表明,供试品溶液中芍药苷色谱峰面积测定结果为257.6,258.3,256.0,257.5,254.6,251.0 mAU·s,相对标准偏差为1.06%;甘草苷峰色谱面积测定结果为75.6,73.9,75.4,73.2,75.5,74.2 mAU·s,相对标准偏差为1.35%,均小于3%,说明该法精密度良好。
2.6 稳定性试验
取1.4中供试品溶液,分别在0,1,2,4,8,12,24 h测定,连续考察24 h,记录色谱峰面积。结果芍药苷色谱峰面积分别为258.2,258.9,257.1,256.2,254.1,248.2 mAU·s,相对标准偏差为1.42%;甘草苷色谱峰面积分别为74.9,74.3,72.8,71.9,73.2,72.6,70.9 mAU*s,相对标准偏差为1.87%,表明供试品溶液在24 h内稳定性良好。
2.7 重复性试验
取批号为150905的百合固金颗粒样品,按照1.4方法制备6份供试品溶液,然后分别注入液相色谱仪进行测定。结果每袋含芍药苷为4.12,4.19,4.10,4.13,4.17,4.13 mg,相对标准偏差为0.81%;甘草苷含量为0.69,0.68,0.70,0.72,0.70,0.71 mg,相对标准偏差为2.02%,表明本方法的重复性良好。
2.8 加样回收试验
精密称取已知含量的百合固金颗粒样品(批号为150905)6份,按1.4方法配制样品溶液。分别精密加入芍药苷储备液3 mL、甘草苷储备液1 mL于样品溶液中。按照方法进行测定,分别计算芍药苷、甘草苷的平均回收率,结果见表2。由表2 可知,芍药苷和甘草苷的回收率分别为96.73%,95.85%,相对标准偏差小于3%,因此该方法具有较好的准确性。
2.9 样品测定
取百合固金颗粒样品,按照1.4方法制备样品溶液,每批样品平行制备2份,按照1.2色谱条件进行测定,以外标法分别计算样品中芍药苷和甘草苷的含量,结果见表3。由表3可知,芍药苷的含量为每袋4.15 mg,符合百合固金颗粒标准中每袋2.1 mg的要求;甘草苷的含量为每袋0.69 mg,虽然含量较低,但用本方法可以测定,由此可见该方法具有一定的适用性。
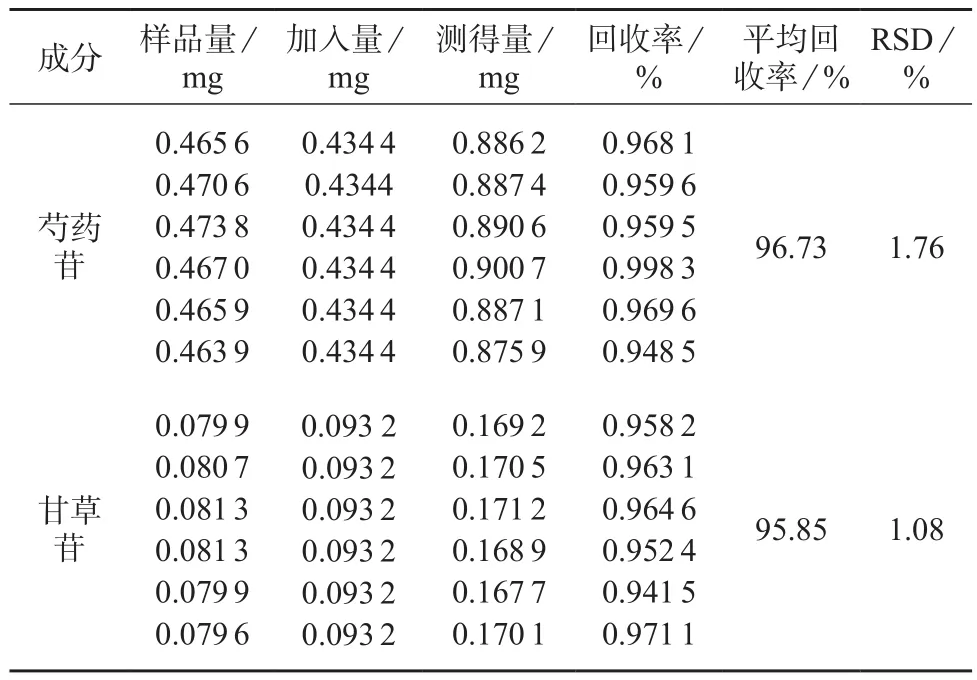
表2 加样回收率试验结果(n=6)

表3 百合固金颗粒样品测定结果(n=2)
3 结语
随着中成药处方中药味的增加,只通过测定其中某一种药物的活性成分来控制其质量,具有很大的缺陷,多组分含量的同时测定能更好地反映中成药的整体质量。采用高效液相色谱法同时测定百合固金颗粒中芍药苷和甘草苷的含量来控制药品质量,比现行标准中单一测定白芍中芍药苷的成分更为有效,方法操作简单,专属性强,可以为更好地评价百合固金颗粒的质量提供参考。
[1] 苏保华.百合固金汤新用[J].陕西中医,2006,27(11): 1 430–1 430.
[2] 马书太. HPLC法测定百合固金颗粒中芍药苷的含量[J].内蒙古中医药,2009,28(6): 37–38.
[3] 董钰明,张军,刘晖,等. RP–HPLC测定百合固金丸中芍药苷的含量[J].中药材,2001,24(5): 362–363.
[4] 尹宁宁,徐华玲,徐丽华.白芍中芍药苷含量测定影响因素的实验研究[J].中国医药导报,2009,6(11): 43–44.
[5] 鲍邢杰,宿树兰.对《中国药典》2010版Ⅰ部中白芍中芍药苷含量测定方法的探讨[J].时珍国医国药,2012,23(5): 1 309–1 310.
[6] 黄山君,杨琪伟,石燕红,等. 一测多评法测定白芍中芍药苷与芍药内酯苷的含量[J].中国中药杂志,2011,36(6): 780–783.
[7] 国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2015:836.
[8] 杜蓉,张孟佑. HPLC法测定加味逍遥丸中芍药苷与甘草苷的含量[J].中国药房,2015,528(18): 2 571–2 572.
[9] 柏冬,范斌,牛晓红,等. HPLC–系统内标法测定桂枝汤中芍药苷、甘草苷、肉桂酸、桂皮醛和甘草酸[J].中草药,2010,41(3): 387–390.
[10] 张科卫,李伟,崔小兵,等. HPLC法测定胃康灵胶囊中芍药苷和甘草苷[J].中草药,2006,37(12): 1 821–1 822.
[11] Gan P,Huang X,Zhong M,et al. Simultaneous determination of eight major constituents in the traditional Chinese medicine Shaoyao-Gancao-Tang by UPLC–PDA[J]. Journal of Medicinal Plants Research,2010,24(4): 2 615–2 621.
[12] 李松,王蓓,浦香兰,等. HPLC–DAD法测定银屑优化颗粒中芍药苷、甘草苷、落新妇苷、迷迭香酸和甘草酸[J].现代药物与临床,2014,29(4): 373–376.
[13] Bai Y,Gong H Y,Li X Q,et al. Rapid Determination of Paeoniflorin and Moisture in Xiaoyao Pills (Concentrated)by Near-Infrared Spectroscopy[J]. Advanced Materials Research,2013,807–809: 1 972–1 977.
[14] 李怡,黄晓虹,林祖文,等. HPLC–MS/MS法测定气滞胃痛片中芍药苷的含量[J].中药材,2013,36(8): 1362–1 364.
[15] 国家药典委员会.中华人民共和国药典.一部[M].北京:中国医药科技出版社,2015:105.
采用碳纳米管调控生活垃圾堆肥重金属释放量的方法
申请公布号:CN106179198A申请公布日:2016.12.07
申请人:天津师范大学
摘要本发明公开了一种采用碳纳米管调控生活垃圾堆肥重金属释放量的方法,淋溶管为高25 cm,内径3 cm的PVC管,管底用纱布封底,每个柱内,底层填充河沙20 g高度1~2 cm,上层填充150 g生活垃圾堆肥和1%(质量分数)的碳纳米材料形成混合基质。模拟夏季暴雨的淋洗,淋溶管静置熟化30天,每天给管内加入适量的蒸馏水,管内土壤含水量为田间持水量,期间,室内温度18~25 ℃,相对湿度35%~65%,光照为透入室内的自然光,第30天,进行淋溶实验,淋溶液用原子吸收分光光度计TAS-990测定其中重金属Cu,Pb,Cd的浓度。实验结果显示:采用碳纳米管调控生活垃圾堆肥重金属释放量的方法可以帮助重金属的原位固定,减少酸雨淋溶量。
一种液相色谱检测三单体的方法
申请公布号:CN106198830A申请公布日:2016.12.07
申请人:泰山医学院
摘要本发明提供一种液相色谱检测三单体的方法,使用FL2200液相色谱仪,N2000色谱工作站,色谱柱Kromstar C18规格为150 mm×4.6 mm,5 µm;流动相a与流动相b的体积比为80~90∶10~20;检测波长:220 nm;流速:0.8 mL/min;进样体积:20 µL;柱温:30℃。使用本发明的检测方法,检测时间小于10 min;分析精度可达0.01 ng;重现性好,相对误差为0.2%;操作简易、分析全部自动化;达到对间苯二甲酸二甲酯-5-磺酸钠的质量进行有效控制的目的。
一种辉光放电质谱法检测金属中痕量杂质元素含量的方法
申请公布号:CN106198712A申请公布日:2016.12.07
申请人:锦州市国家光伏材料质量监督检验中心
摘要本发明提供了一种辉光放电质谱法检测金属中痕量杂质元素含量的方法。该方法包括以下步骤:(1)将金属样品加工成片状或针状,置于硝酸溶液中进行超声清洗,烘干;(2)将辉光放电质谱仪的样品仓抽真空,打开球阀,将金属样品推入离子源腔内,打开高压和辉光,通入工作气体,调节放电电压和放电电流,通过离子源调谐将分辨率调到3 000~4 800,基体峰强度为0.05~0.25 V,预溅射30~80 min;(3)选择金属基体和需测定的杂质元素,进行直流辉光放电质谱分析,得到金属痕量杂质元素的浓度。本发明无需研磨以及抛光按规格切割好的样品,只需清洗样品,预处理简单,能有效避免污染;固体直接进样,操作简单易行;对离子源进行有效调谐,灵敏度高,检测限低,分辨率高,可一次性分析主含量元素至1×10–12级的元素。
一种超临界流体色谱–气相色谱–质谱测定卷烟主流烟气中苯并[a]芘的方法
申请公布号:CN106198796A申请公布日:2016.12.07
申请人:云南中烟工业有限责任公司
摘要本发明公开了一种超临界流体色谱(SFC)–气相色谱–质谱(GC–MS)测定卷烟主流烟气中苯并[a]芘的方法。本方法是在抽吸卷烟后,向捕集卷烟主流烟气总粒相物的滤片中加入内标物及环己烷溶液,超声提取后,萃取液用超临界流体色谱进行分离净化,切割含有苯并[a]芘的馏分再用GC–MS分析,根据目标物峰和同位素内标物峰的面积比定量。本发明能实现复杂基体中痕量待测成分与干扰物质的分离,排除了其它物质的干扰,提高了检测的精确性,具有前处理简单、分离速度快,定量准确、检出限低等优点。
锂电池电解液的分析方法
申请公布号:CN106198813A申请公布日:2016.12.07
申请人:凯思普科技有限责任公司
摘要本发明涉及锂电池电解液的分析方法,锂电池电解液至少包括锂盐,锂盐包括至少一种含硼锂盐,该分析方法包括以下步骤:将待分析的电解液与甲醇水溶液混合,得到混合液;加热混合液至40~80℃,搅拌并保持8~24 h;停止搅拌,静置分层,分离出有机层;使用气相色谱仪分析有机层中硼酸酯化物的含量;计算得到电解液中硼元素的含量。本发明能够快捷方便地得到电解液中含硼锂盐的定性定量数据。
Determination of Paeoniforin and Liquoritin in Baiheguji Granules by HPLC
Zhang Xin, Wang Yunxia
(Xi’an Institute for Food and Drug Control, Xi’an 710054, China)
A method was established for the simultaneous determination of paeoniflorin and liquoritin content in Baiheguji granules by HPLC. Welch UltimateXB–C18colum (250 mm×4.6 mm,5 µm) was adopted with acetonitrile–0.1% phosphoric acid (14∶86) as the mobile phase at fow rate of 1.0 mL/min. Detection wave length was 230 nm,colum temperature was 30℃. The content of Paeoniforin and Liquoritin was linear with the peak area in the range of 14.91–149.12 ng and 34.75–347.52 ng, respectively, and the correlation coeffcient was 0.999 5, 0.999 2, respectively. The average recovery were 96.7%,95.9%, respectivly, and the relative standard deviation of detection results was less than with 3%(n=6). The method has advantages of simple operation,high accuracy stability and good reproducibility. It can be used for quality control of Baiheguji granules effectively.
Baiheguji; granules; paeoniforin liquoritin; content determination; HPLC
O657.7
:A
:1008–6145(2017)01–0088–04
10.3969/j.issn.1008–6145.2017.01.023
联系人:张鑫;E-mail:awoshizx@126.com
2016–10–20
