QuEChERS前处理联合UPLC–MS/MS法检测花生中22种农药残留
2017-02-16李婵君王毅红王彦超董素静段鹿梅
李婵君,王毅红,王彦超,董素静,段鹿梅
(郑州市农产品质量检测流通中心,郑州 450006)
QuEChERS前处理联合UPLC–MS/MS法检测花生中22种农药残留
李婵君,王毅红,王彦超,董素静,段鹿梅
(郑州市农产品质量检测流通中心,郑州 450006)
建立了QuEChERS(Quick,Easy,Cheap,Effective,Rugged,Safe)前处理联合UPLC–MS/MS法检测花生中22种农药残留的方法。样品用10 mL乙腈提取,以多壁碳纳米管、N-丙基乙二胺为吸附剂,对2 mL提取液进行净化,净化液稀释至2倍体积,以MRM扫描方式、正负离子模式同时分析。22种农药在10,20,50 μg/kg 3个添加水平下,平均回收率为70.6%~121.2%,相对标准偏差小于10%(n=6);多菌灵、抗蚜威、扑草净在0.05~10μg/L,啶虫脒、氟虫腈砜、苯醚甲环唑、哒螨灵、嘧霉胺、嘧菌酯在0.5~20 μg/L,烯酰吗啉、噻虫嗪、氟啶脲、灭幼脲、吡虫啉、甲维盐、除虫脲、氟虫腈、氟甲腈、氟虫腈亚砜、咪鲜胺、二甲戊灵在0.5~50 μg/L之间,阿维菌素在0.5~100 μg/L范围内线性良好,相关系数r2均大于0.995 0。22种农药的定量限在2 μg/kg以下,远低于各待测农药最高残留限量标准(MRL)。该法适于花生中农药残留的同时快速检测。
QuEChERS;超高液相串联质谱法;花生;农药残留
花生是我国重要的粮油作物之一,我国是世界上花生总产量和花生油总产量最大的国家,同时,我国也是世界第一花生出口大国。随着国际花生贸易的发展,工业发达国家越来越将农药残留作为限制花生出口的技术性贸易壁垒,同时更加严格地规定了对进口花生的检验检疫标准,这些规定对我国花生出口产生了较大压力,也对我国花生中农药残留的检测技术提出了挑战。
花生中含有大量油脂,同时富含高蛋白和色素,这些物质如果在样品前处理过程中不能去除完全,将给后续的农药多残留检测造成很多困难[1–2]。我国尚未对花生中的农药多残留分析形成统一的标准[3–4],花生中的农药多残留分析要参考多个标准的相关项目,这些不同的标准不仅前处理方法不尽相同,而且检测所需的分析仪器也不同,涉及到的样品前处理和分析方法有分散固相萃取[5]、基质固相分散辅助加速溶剂萃取、凝胶渗透色谱、液相色谱、气相色谱以及色谱–质谱联用等[5–10]。另外,传统的分析方法操作复杂,耗时较长,很难满足对农药多残留高效快速筛查与分析的需求。
近年来,以石墨稀为结构单元的碳纳米管的开发与应用飞速发展,得到了各国研究者的高度关注。因其直径为纳米级,比表面积大,扩散距离短,对多种有机化合物具有良好的吸附性,只需使用少量的吸附剂即可在较短的平衡时间内实现萃取分离,因此碳纳米管被广泛用于各种药物分析中[11–18]。另外,碳纳米管还可以与传统的固相萃取净化材料如石墨化碳黑(GCB)、N-丙基乙二胺(PSA)、十八烷基硅胶(C18)等混合使用,实现更显著的净化效果。
笔者用10 mL乙腈提取花生样品,以多壁碳纳米管、PSA为吸附剂对2 mL提取液进行净化,净化液稀释至2倍体积,使用Waters Xevo TQ–S液质联用仪,6 min内完成22种农药上机分析。采用基质加标法进行定量,该方法消除花生基质干扰的效果好,分析快速高效,灵敏度高,为花生样品中农药残留检测提供新思路。
1 实验部分
1.1 主要仪器与试剂
超高压液相色谱串联质谱仪:Waters Xevo TQ–S型,美国Waters公司;
涡旋混合器:Hedolph Multi Reax型,德国海尔道夫公司;
高速冷冻离心机:SIGMA 3K15型,德国Sigma公司;
纯水机:Milli-Q Advantage A10型,法国Millipore公司;
震荡器:SHA–C型,金坛市杰瑞尔电器有限公司;
无水硫酸镁:分析纯,使用前于650℃下灼烧4 h,天津市化学试剂三厂;
氯化钠:分析纯,使用前于650℃下灼烧2 h,天津市科密欧化学试剂有限公司;
乙腈、甲醇:色谱纯,美国Merck公司;
甲酸:色谱纯,美国Tedia公司;
多壁碳纳米管(MWCNTs):直径大于50 nm,长度为10~20 µm,纯度为98%,比表面积为40 m2/g,南京先丰科技有限公司;
GCB,PSA,C18:美国Agilent公司;
农药标准品:分别为烯酰吗啉、噻虫嗪、氟啶脲、灭幼脲、啶虫脒、吡虫啉、多菌灵、甲维盐、阿维菌素、除虫脲、氟虫腈、氟甲腈、氟虫腈亚砜、氟虫腈砜、苯醚甲环唑、哒螨灵、嘧霉胺、抗蚜威、咪鲜胺、嘧菌酯、二甲戊灵、扑草净,质量浓度均为1 000 mg/L,农业部环境保护科学监测所;
单标准储备液:100 mg/mL,准确吸取1 mL各农药标准品于10 mL容量瓶中,噻虫嗪、多菌灵、吡虫啉、啶虫脒、烯酰吗啉、除虫脲、灭幼脲、甲维盐、氟啶脲、阿维菌素、哒螨灵用甲醇定容,抗蚜威、嘧霉胺、嘧菌酯、扑草净、咪鲜胺、氟虫腈、苯醚甲环唑、氟甲腈、氟虫腈砜、氟虫腈亚砜、二甲戊灵用丙酮定容,于–18℃冰箱中密封储存备用,根据需要用溶液稀释成相应浓度的单标准工作溶液;
混合标准中间液:分别移取一定量的单标准储备液于10 mL容量瓶中,用乙腈定容得到混合标准中间液,于–18℃冰箱中密封储存备用,根据需要用乙腈稀释为系列浓度的混合标准工作液;
花生样品:采样于农贸市场、超市等流通环节,经粉碎机粉碎后备用;
实验用水为一级水,用Millipore纯水机制得。
1.2 仪器工作条件
1.2.1 液相色谱
色谱柱:ACQUITY–UPLC–BEH C18柱(50 mm×2.1 mm,1.7µm,美国Waters公司);流动相:A为乙腈,B为5 mmol/L乙酸铵–0.02%甲酸水溶液,流量为0.3 mL/min,梯度洗脱程序见表1;柱温:30℃;进样体积:5 µL。
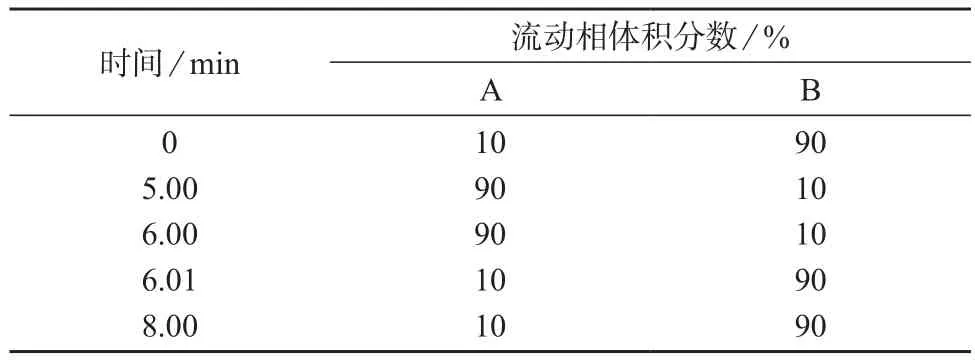
表1 流动相梯度洗脱程序
1.2.2 质谱
离子源:电喷雾离子源;扫描方式:正离子扫描;检测方式:多反应监测(MRM);毛细管电压:正离子模式为3.0 kV,负离子模式为2.0 kV;离子源温度:150℃;脱溶剂温度:500℃;脱溶剂气流量:1 000 L/h;锥孔气流量:50 L/h;载气:氮气;碰撞气体:氩气。
22种农药的总离子流图见图1,质谱采集参数见表2。

图1 22种农药的总离子流图
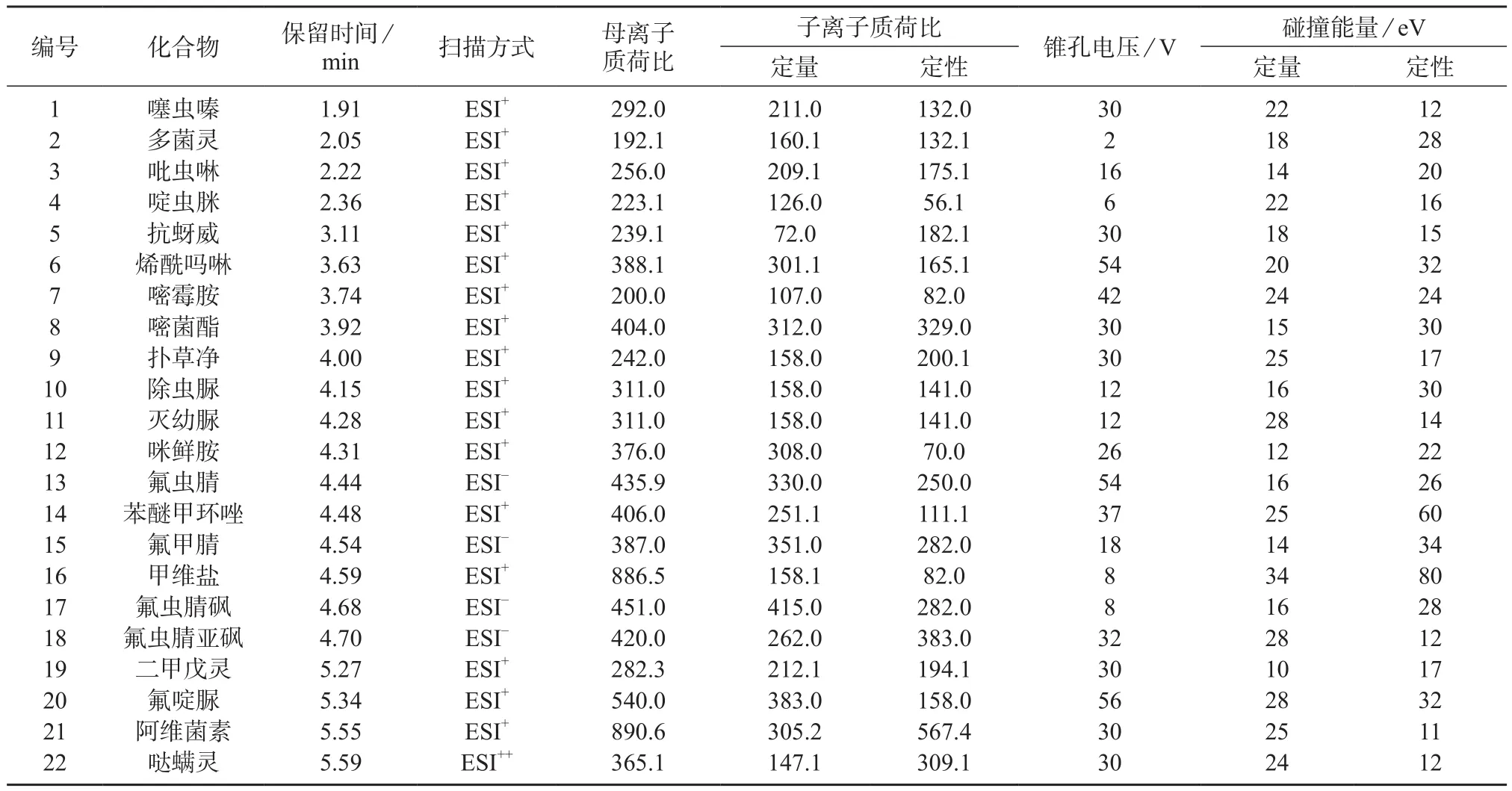
表2 22种农药的质谱采集参数
1.3 实验方法
称取匀浆后的5.0 g样品于带盖的聚丙稀离心管中,加入超纯水5 mL,涡旋使水充分浸润样品,静置20 min,加入乙腈溶液10 mL,振荡15 min,再加入1 g NaCl和4 g无水MgSO4,振荡30 s,以5 000 r/min冷冻离心5 min。吸取离心后所得上层清液2.0 mL于预先加入300 mg无水硫酸镁、50 mg PSA和10 mg MWCNTs的离心管中,立即涡旋30 s,然后以8 000 r/min冷冻离心5 min。吸取上清液0.50 mL,加入0.50 mL超纯水,混匀后过0.22 µm滤膜,以UPLC–MSMS法测定。
2 结果与讨论
2.1 流动相的优化
试验考察22种农药在乙腈–水、甲醇–水流动相体系中的分离度、色谱峰形及响应值,结果表明绝大多数化合物在乙腈–水体系中获得较好的色谱峰。与甲醇相比,乙腈黏度较小,系统压力较低,所以选择乙腈–水体系作为流动相。在水溶液中加入乙酸铵,分别用2 mmol/L乙酸铵–0.02%甲酸、5 mmol/L乙酸铵–0.02%甲酸、2 mmol/L乙酸铵–0.1%甲酸、5 mmol/L乙酸铵–0.1%甲酸为流动相分析化合物,发现随着甲酸比例减少、乙酸铵浓度的增加,大部分化合物的离子化效率、目标物的响应值都有提高,特别是多菌灵的色谱峰形有明显改善,因此选择5 mmol/L乙酸铵–0.02%甲酸水溶液和乙腈为流动相。
2.2 样品浸泡对回收率的影响
花生样品中含有较多的油脂,加入乙腈提取时,样品不易分散开,影响目标物的提取效率。试验考察了不加水与加水浸泡提取对样品中目标物回收率的影响,当加水量与样品量比例为1∶1(质量比)时,实现样品的完全浸润并分散。实验结果表明,与样品前处理过程加水提取相比,在不加水提取时,大部分目标物回收率无显著差别,而多菌灵、吡虫啉回收率有较大变化,回收率从不加水时的30%,70%分别提高到加水后的约80%,100%。根据以上试验结果,样品前处理过程选择按样品比例1∶1加水的提取方式。
2.3 吸附剂组合用量的优化
QuEChERS(Quick,Easy,Cheap,Effective,Rugged,Safe)方法的原理是利用净化剂与样品基质中干扰物质的相互作用吸附杂质,从而净化基质。传统QuEChERS方法使用较多的净化剂材料有PSA,C18,GCB等。本实验采用多壁碳纳米管MWCNTs和传统净化材料组合:MWCNTs/PSA/MgSO4,C18/PSA/MgSO4,GCB/PSA/MgSO4,比较其净化效果,称取5 g空白花生样品并添加0.5 µg/mL混合标准工作液0.2 mL,按照1.3方法提取。提取液分别用以上3组吸附剂组合进行净化处理。从净化后定容溶液的混浊度来看,MWCNTs/PSA/MgSO4组合净化后的提取液最为澄清,净化效果最好;其次为C18/PSA/MgSO4组合;GCB/PSA/MgSO4组合净化后的提取液最为混浊,脂肪含量较多,净化效果最差。使用基质匹配标准工作液制备工作曲线进行校正,得到不同吸附剂处理样品处理后各目标物的加标回收率,结果表明MWCNTs/PSA/MgSO4组合的回收率与C18/PSA/MgSO4组合的回收率相差不大,但明显优于GCB/PSA/MgSO4组合的回收率。综合不同类型吸附剂组合的净化效果和加标回收率,最终选择MWCNTs/PSA/MgSO4作为前处理过程的净化吸附剂。
2.4 标准工作曲线和定量限
采用基质匹配标准工作溶液制定标准工作曲线。根据各化合物不同的灵敏度选定合适的标准曲线浓度范围,分别对各系列标准溶液进行UPLC–MSMS分析,以各组分色谱峰面积(A)对质量浓度(c,ng/mL)进行线性回归,22种分析物在不同的浓度范围内线性关系良好,相关系数(r2)均大于0.995 0,化合物定量限(LOQ,S/N=10)在0.01~2.0 µg/kg之间,满足欧盟和日本等规定的最大残留限量要求,具体数据见表3。
2.5 加标回收试验
在最优实验条件下,绘制了花生基质匹配标准工作曲线,对22种农药在花生样品中的加标回收率进行考察。在10,20,50 µg/kg 3个加标浓度水平下,每个浓度水平重复测定6次,测定结果见表4。由表4可知,22种农药的平均回收率在70.6%~121.2%之间,相对标准偏差为0.88%~9.3%(n=6)。回收率和精密度均满足农药残留检测要求。
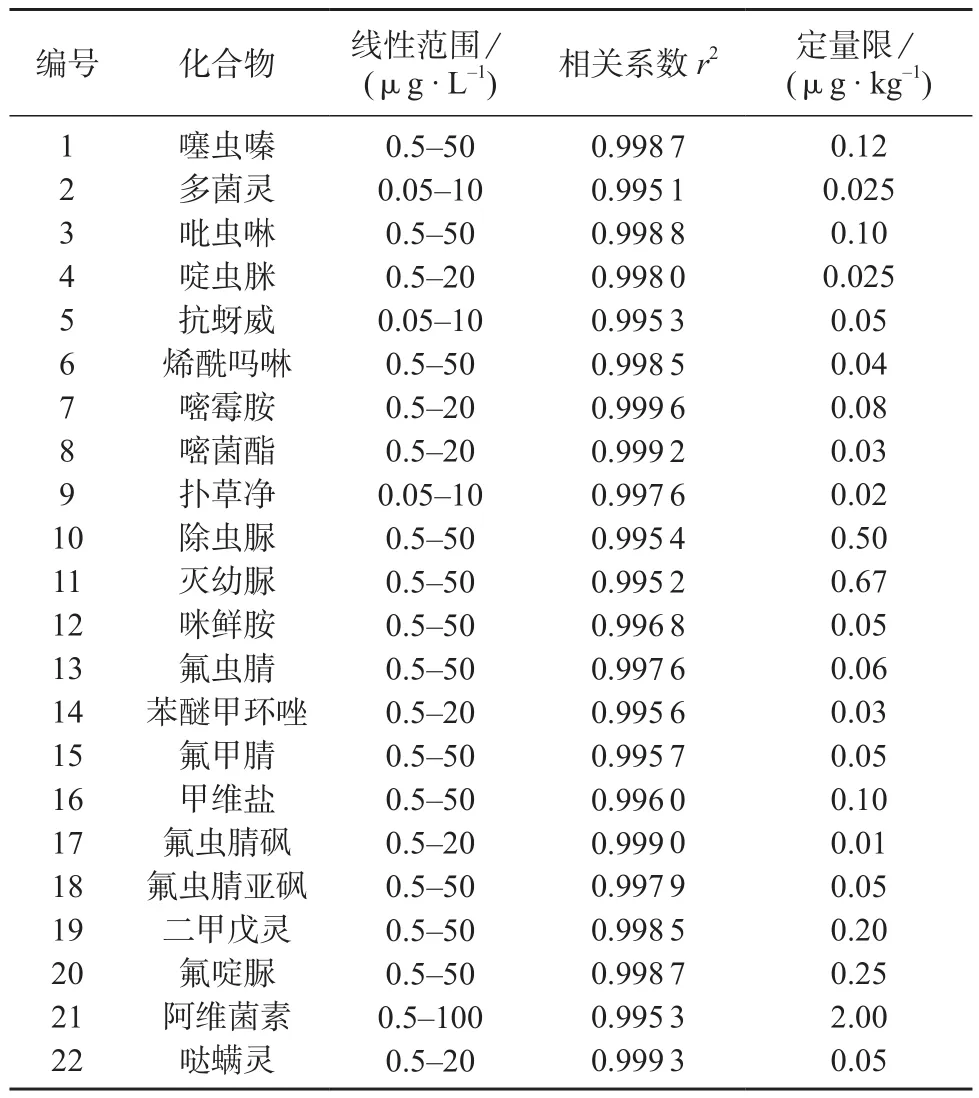
表3 22种农药的线性范围、相关系数、定量限
3 结语
通过优化实验条件建立了QuEChERS前处理方法联合液相色谱–质谱联用技术快速检测花生中22种农药残留的方法。采用乙腈提取,以PSA,MWCNTs作为吸附对样品进行净化,定容后进行UPLC–MS/MS检测。此方法简化了样品前处理步骤,节约了分析时间,有机溶剂用量少,具有较好的准确度、较高的灵敏度与回收率,能有效净化花生中的杂质,定量限能满足目前国内外花生中相应药物残留检测的要求,实现了对花生中农药多残留的同时分析,是一种经济、环保的样品前处理与分析技术。
[1] 郑永权.农药残留研究进展与展望[J].植物保护,2013,39(5): 90–98.
[2] 董永辉,孟金祥,宋利伟.花生病虫害发生及防治[J].陕西农业科学,2010(4): 220–221.
[3] 刘若微,姚晗珺,董国堃,等.我国输日花生农残超标问题分析[J].花生学报,2007,36(4): 7–11.
[4] 丁小霞,李培武,周海燕,等.花生农药最大残留限量标准比对研究[J].中国油料作物学报,2011,33(5): 527–531.
[5] 马金凤,刘雪,赵志强,等.凝胶渗透色谱–气相色谱质谱法测定花生中6种除草剂农药残留[J].化学分析计量,2012,21(4): 43–46.
[6] Angelika M W,Marek B. Rapid method for the determination of organ chlorine pesticides and PCBs in fish muscle samples bymicrowave-assisted extraction and analysis of extracts by GC–ECD[J]. J AOAC In,2010,93(6): 1 987–1 994.
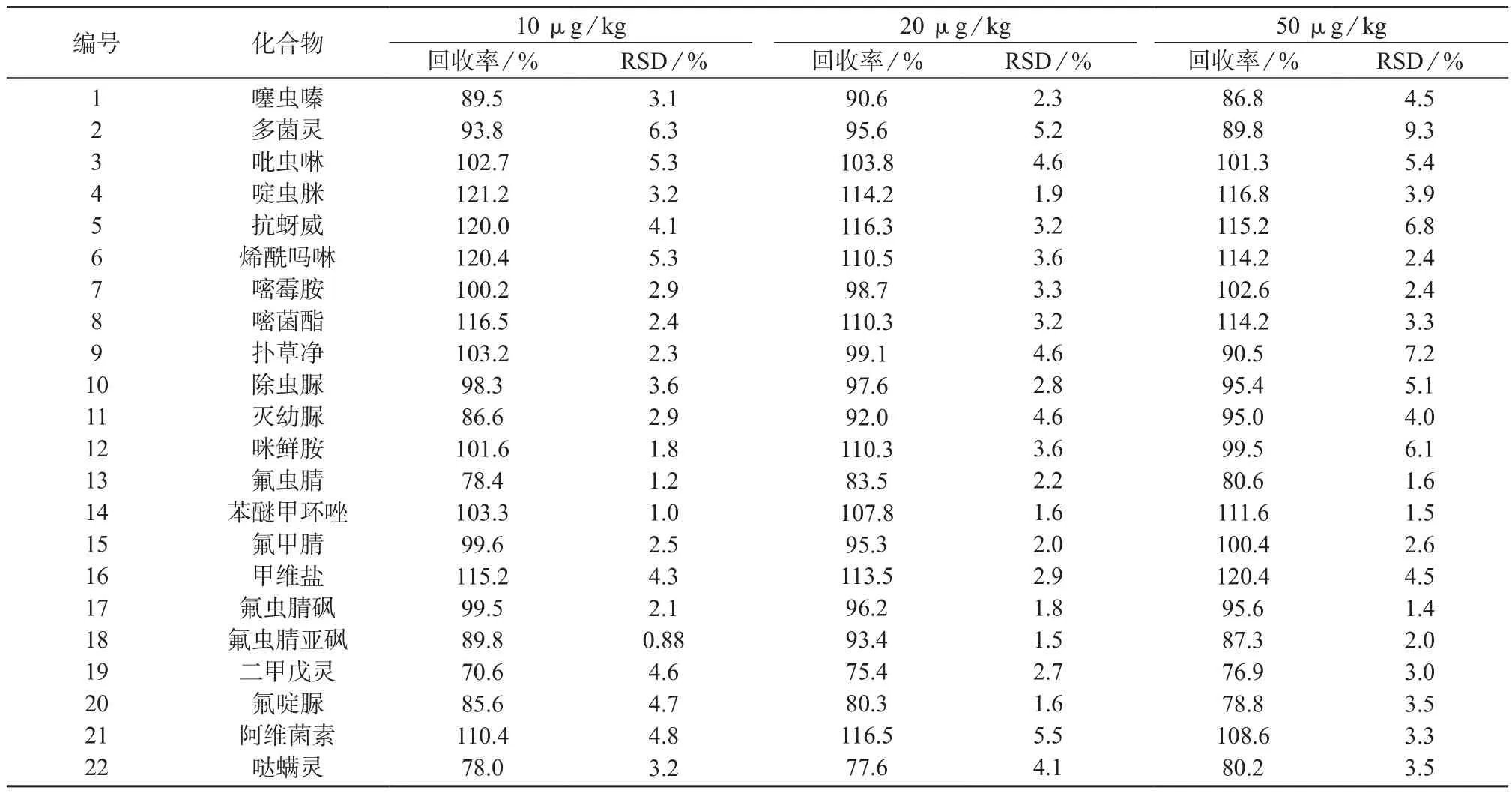
表4 加标回收试验结果(n=6)
[7] Liu Y H,Shen D Y,Mo R H,et al. Simultaneous determination of 15 multiresidue organophosphorous pesticides in camellia oil by MSPD–GC–MS[ J]. Bulletin of Environmental Contamination and Toxicology,2013,90(3): 274–279.
[8] 刘芳,马琳.分散固相萃取–气相色谱法测定花生及大豆中5种除草剂残留[J].现代农药,2016,15(2): 39–41.
[9] 马金凤.凝胶渗透色谱–气相色谱质谱法测定花生中46种农药残留[D].山东农业大学,2012.
[10] 于辉,史俊稳,赵萍.基质固相分散辅助加速溶剂萃取–气相色谱法测定花生中有机磷农药残留[J].食品科学,2010,31(22): 427–430.
[11] 张素玲,杜卓,李攻科.碳纳米管在样品前处理中的应用[J].化学通报,2011,74 (3): 201–207.
[12] 吴新华,丁利,李忠海,等.多壁碳纳米管固相萃取–高效液相色谱–串联质谱法测定食品接触材料中双酚–二环氧甘油醚的迁移量[J].色谱,2010,28(11): 1 094–1 098.
[13] 易谷洋,魏瑞萍,谭小旺,等,多壁碳纳米管固相萃取–高效液相色谱法测定水中四溴双酚A[J].理化检验:化学分册,2013(4): 487–488.
[14] 寇立娟,梁荣宁.羧基化多壁碳纳米管固相萃取–液相色谱–串联质谱法测定环境水体中四溴双酚A和双酚A[J].色谱,2014,32(8): 817–821.
[15] 刘建林,张琛,王夏娇.基于碳纳米管的固相萃取–分散液液微萃取测定水中多种痕量环境雌激素[J].高等学校化学学报,2012,33(1): 37–43.
[16] 曹慧,陈小珍,朱岩,等.多壁碳纳米管固相萃取技术同时测定蜂蜜中多类兽药残留[J].高等学校化学学报,2013,34(12): 2 710–2 715.
[17] 马立利,贾丽,周欣燃,等,多壁碳纳米管滤过型净化柱净化–超高效液相色谱/串联质谱法同时测定生姜中的涕灭威及其代谢物[J].色谱,2014(6): 635–639.
[18] 赵海香,刘海萍,闫早婴.多壁碳纳米管固相萃取净化–高效液相色谱法测定猪肉和鸡肉中的磺胺多残留[J].色谱,2014(3): 294–298.
Determination of 22 Pesticide Residues in Peanut by QuEChERS–UPLC–MS/MS
Li Chanjun, Wang Yihong, Wang Yanchao, Dong Sujing, Duan Lumei
(Zhengzhou Testing Center for Quality of Agricultural Products, Zhengzhou 450006, China)
QuEChERS–UPLC–MS/MS was proposed for determination of 22 pesticide residues in peanut. The sample was extracted with 10 mL acetonitrile. 2 mL of the supernatant was taken and cleaned up with multi-walled carbon nanotubes (MWCNTs) and PSA. After being diluted to 2 times volume, the purifed solution was determined by UPLC–MS/MS under the mode of MRM. ESI was adopted in MS operated in the positive negative ion switch mode. Recovery rates at the three concentrations of 10,20,50 µg/kg ranged from 70.6% to 121.2% with RSDs less than 10%(n=6). Linear relationships between values of peak area and mass concentration were found in the range of 0.05–10 µg/L for carbendazim, pirimicarb and prometryne, 0.5–20 µg/L for acetamiprid, fpronil-sulfone, difenocoazole,pyridaben, pyrimethanil and azoxystrobin, 0.5–50 µg/L for dimethomorph, thiamethoxam, chlorfuazuron, chlorbenzuron, imidacloprid, emamectin benzoate, difubenzuron, fpronil, fpronil-desulfnyl, fpronil-sulfde, prochloraz and pendimethalin, and 0.5–100 µg/L for abamectin, with the linear relative coeffcients (r2) more than 0.995 0. The LOQs of 22 pesticide residues were less than 2 µg/kg, which were signifcantly lower than the respective MRLs. This method can be used in the determination of 22 pesticide residues in peanut.
QuEChERS; UPLC–MS/MS; peanut; pesticide residues
O657.7
:A
:1008–6145(2017)01–0033–05
10.3969/j.issn.1008–6145.2017.01.008
联系人:李婵君;E-mail: chanjunli@163.com
2016–11–24
