液相色谱-串联质谱法快速筛查减肥类中药及保健品中非法添加30种化学药
2017-02-15吴惠勤朱志鑫黄晓兰罗辉泰林晓珊马立果钟巧丽蒋娅兰
黄 芳,吴惠勤,朱志鑫,黄晓兰,罗辉泰,林晓珊,马立果,钟巧丽,蒋娅兰
(中国广州分析测试中心 广东省分析测试技术公共实验室,广东 广州 510070)
研究报告
液相色谱-串联质谱法快速筛查减肥类中药及保健品中非法添加30种化学药
黄 芳*,吴惠勤,朱志鑫,黄晓兰,罗辉泰,林晓珊,马立果,钟巧丽,蒋娅兰
(中国广州分析测试中心 广东省分析测试技术公共实验室,广东 广州 510070)
建立了中药及保健品中30种减肥类化学药的高效液相色谱-串联质谱(LC-MS/MS)快速筛查方法。实验优化了前处理方法、色谱分离条件和质谱参数。样品采用甲醇超声萃取,Agilent poroshell 120 EC-C18(4.6 mm×100 mm,2.7 μm)色谱柱分离,流动相为乙腈-水溶液(含10 mmol/L甲酸铵,0.1%甲酸,10%甲醇),梯度洗脱,流速0.25 mL/min,采用正负离子切换模式的电喷雾质谱检测,多反应选择离子监测(MRM)。该方法能同时筛查30种化学药,覆盖面广,简便、快速、准确可靠,已用于减肥类中药及保健品中非法添加化学药的筛查及检测。
减肥类化学药;液相色谱-串联质谱;中药及保健品
随着社会的发展,人们物质生活越来越丰富,肥胖人群增多,加上人们对审美观念的变化,使得减肥成为现代社会的热门话题。面对减肥,多数人会选择相对轻松的吃药方式,认为吃药可以代替减肥运动和饮食控制。由此市场上出现了各式各样的减肥药产品,为达到见效快的目的,有些产品打着纯中药制剂的旗号,实际在其中添加了具有减肥或辅助减肥效果的化学药,例如有食欲抑制剂西布曲明、泻药酚酞、利尿剂噻嗪酮、降糖药二甲双胍等,而消费者在不知情的情况下长期服用这些产品,会对身体产生极大伤害。由于在复杂中草药体系中添加化学药具有隐蔽性且不易检出的特征,近年来虽然相关部门加大力度对市场抽检以及风险评估等监管活动,但见效不佳,非法添加行为仍屡禁不止[1-2]。
针对减肥类药物的检测,我国现行的标准有药品补充检验标准[3],其中规定了非法添加的3项减肥类化学药西布曲明、麻黄碱、芬氟拉明的检测方法,而实际检验发现,除了含有这3种成分,另有部分产品还含有其它功效类似的降糖药、利尿药和降脂药。因此采用现有的标准不能达到有效监管目的,需要建立品种更多、覆盖面更广的多成分同时筛查方法作技术支撑。近年来减肥药物的检测方法有高效液相色谱法[4-5],但此法只能通过保留时间及紫外吸收光谱定性,不能确证化合物的结构信息,需要结合质谱确证才能用于实际工作中;而现有报道最多的是采用灵敏度高、选择性好的液相色谱-质谱法(HPLC-MS/MS)[6-8],其中王柯等[8]建立了HPLC-MS/MS测定28种减肥类药物的分析方法,是所有报道中药物种类最多的方法。近年来本课题组一直在研究该类物质的筛查检测方法[9],在筛查品种以及检测技术上有了较多的积累,现建立了能同时检出30种减肥类化学药的快速筛查方法。这30种物质均是实际检测工作中被检测或剖析出来的成分,如西布曲明和酚酞的被检出频率最高;氟西汀是从实际样品中通过剖析鉴定出的成分;氯卡色林是2012年美国新批准上市的新一代减肥药[10],检测方法尚无文献报道。本文所建立的方法有以下优势:①能同时筛查30种化学药,覆盖面广,对有些天然药物如大黄素、咖啡因、茶碱等因不易判断是否非法添加而不列入本方法,方法更实用;②氯卡色林和氯贝丁酯为文献尚未报道的非法添加成分;③采用正、负离子切换模式采集数据,提高了氯噻嗪、氢氯噻嗪、噻呋米和氯噻酮的灵敏度,达到同时测定的目的。本方法已经应用于实际样品的检测,为相关监管部门提供了技术支撑,可为此类非法添加的风险评估提供服务。
1 实验部分
1.1 仪器与试剂
Agilent 1200 SL Series RRLC /6410B Triple Quard MS 快速高效液相色谱-串联四极杆质谱联用仪( 美国安捷伦公司);赛多利斯TP-114电子天平(美国Sartorious公司);KQ2200型台式机械超声波清洗器(东莞市超声波设备有限公司)。甲醇、乙腈(色谱纯,德国Merk公司),乙酸铵(阿拉丁试剂),实验用水为二次蒸馏水,其余试剂均为分析纯。
30种标准品:二甲双胍、阿卡波糖、氯噻嗪、苯乙双胍、氢氯噻嗪、罗格列酮、氯卡色林、氯噻酮、芬氟拉明、吡格列酮、噻呋米、氟西汀、酚酞、去甲基西布曲明(东布曲明)、西布曲明、格列吡嗪、甲苯磺丁脲、阿托伐他汀、格列齐特、比沙可啶、格列苯脲、格列美脲、格列喹酮、氯贝丁酯、瑞格列奈、洛伐他汀、辛伐他汀、利莫那班、非诺贝特、奥利司他,纯度均大于 95.2%,均购自中国食品药品检定研究院和德国 Dr.Ehrenstorfer 公司。
1.2 标准溶液配制
分别准确称各标准品5.00 mg,用甲醇(二甲双胍和阿卡波糖用水)超声溶解并定容至10 mL,得到500 mg/L的标准储备液,临用时以基质提取液将上述标准溶液稀释为系列混合标准工作液。
1.3 样品前处理
片剂去除糖衣部分,磨成细粉,胶囊取内容物,准确称取0.1 g,置于10 mL容量瓶中,加甲醇8 mL,超声处理20 min,甲醇定容至刻度,过0.20 μm微孔滤膜,待测。
1.4 液相色谱-质谱条件
色谱柱:Agilent poroshell 120 EC-C18(4.6 mm×100 mm,2.7 μm),流动相:A为乙腈,B为水溶液(含有10 mmol/L甲酸铵,0.1%甲酸,10%甲醇),梯度洗脱程序:0~2 min, 100% B;2~3 min,100%~75% B;3~5 min, 75%~20% B;5~8 min, 20%~0% B;8~10 min, 0% B;10~11 min, 0%~100% B;11~14 min, 100% B。流速0.25 mL/min,柱温30 ℃,进样量2 μL。
离子源为电喷雾(ESI)离子源,干燥气(N2)温度350 ℃,雾化气(N2)压力275.8 kPa,干燥气(N2)流量10 L/min,电喷雾电压4 000 V,扫描方式为多反应监测(MRM)模式,采用正负离子切换方式采集,优化后的质谱分析参数见表1。

表1 30种减肥化学药的质谱分析参数
*quantitative ion
2 结果与讨论
2.1 样品前处理条件的优化
二甲双胍和阿卡波糖均在水中易溶,在甲醇中溶解,在乙醇中微溶,阿卡波糖在乙腈中不溶,其余各药物均在甲醇中溶解。为了获得较好的回收率和除杂效果,实验对比了纯甲醇、80%甲醇、60%甲醇3种提取溶剂的提取效果,结果表明,纯甲醇作提取剂时每个成分能得到理想的回收率,且滤液洁净,无需进一步净化,简化了样品前处理过程,因此采用甲醇作为提取溶剂。
2.2 质谱条件的优化
分别将30种化学药的标准溶液注入质谱仪进行参数优化,获得相应的[M+H]+和[M-H]-准分子离子峰,然后针对母离子作二级子离子全扫描,获得碎片离子信息,选择丰度较高的2个碎片离子作特征离子,优化碰撞能量使其响应最大,最后得到该化合物的2个MRM离子对及相应的质谱参数。
由于不同物质的化学性质各异,在电喷雾离子源中的离子化程度不同,优化参数时需采用不同浓度进样,才能得到理想参数。化合物氯贝丁酯不易离子化,其浓度在10 mg/L以下不能得到分子离子峰,改用较大浓度100 mg/L进样,才能得到其分子离子峰m/z243+,氯噻嗪、氯噻酮、噻呋米和氢氯噻嗪在理论上采用正、负离子检测均可,因此实验分别优化得到其正、负离子的MRM参数。
为得到每种物质的理想灵敏度,实验对比采用正离子(MRM)模式和正、负离子切换采集MRM模式[12]下氯噻嗪、氯噻酮、噻呋米和氢氯噻嗪的检出限,结果显示,正离子模式下氯噻嗪和氯噻酮的检出限为5 mg/kg,噻呋米和氢氯噻嗪为10 mg/kg,而正负离子切换模式下能得到更低的检出限,其中氯噻嗪、氢氯噻嗪和噻呋米为0.01 mg/kg;氯噻酮为0.05 mg/kg(见表2 ),比正离子模式低1~2个数量级,大大增加了此类物质的灵敏度。因此,本实验选用正负离子切换模式的MRM模式采集数据。
2.3 色谱条件的优化
各种化学药的理化性质差别大,在色谱柱上保留特征也不同,为得到理想的色谱峰及合适的分离度,对比选择了性质不同的4种色谱柱,考察其保留特征及分离情况。4种色谱柱分别为A:Agilent Poroshell 120 EC-C18(3.0 mm×100 mm,2.7 μm);B:Agilent Poroshell 120 Phenyl Hexyl(2.1 mm×100 mm,2.7 μm);C:Agilent Poroshell 120 PFP(2.1 mm×100 mm,2.7 μm);D:Agilent Poroshell 120 EC-C18(4.6 mm×50 mm,2.7 μm)。研究结果表明:使用B柱时,阿卡波糖的保留时间短,保留时间为1.61,2.08 min,二甲双胍出峰时间为1.3 min,几乎没有保留而与溶剂同流出;使用C柱时,阿卡波糖的出峰时间为1.5 min几乎无保留,另有芬氟拉明和氯卡色林的峰形拖尾严重;使用D柱时,阿卡波糖的拖尾严重,各种物质相对分离不如A柱理想。由于采用A柱时各色谱峰分离比较理想,峰形对称,所以选择A柱作为分离柱。

图1 30种化学药混合标准溶液的总离子流色谱图Fig.1 TIC chromatogram of 30 compounds mixed standard solution
流动相对比了甲醇-水和乙腈-水混合溶剂,并分别添加甲酸铵和甲酸考察其对30种化合物的色谱行为和离子化程度。结果显示,加入甲酸会提高正离子物质的灵敏度,加入甲酸铵可提高负离子物质的灵敏度。若只加入甲酸铵,会导致西布曲明的峰严重展宽,几乎得不到明显色谱峰,而加入甲酸后峰形明显改善,检出限降低2~3个数量级。虽然加入甲酸后导致阿卡波糖分裂为两个峰,但两个峰均峰形尖锐,不影响其检出限和定量的准确性;另外在水系中加入10%甲醇,既能改善各物质的分离,又能防止流动相产生微生物或沉淀导致柱压升高。因此流动相体系采用乙腈-甲酸和甲酸铵水溶液,水溶液中含有10%甲醇,梯度洗脱。
综合以上各因素,得到最佳色谱条件为:色谱柱为Agilent Poroshell 120 EC-C18(3.0 mm×100 mm,2.7 μm);流动相:A为乙腈,B为水溶液(含有10 mmol/L甲酸铵、0.1%甲酸、10%甲醇),此条件下所得各种化合物的总离子流图和定量特征离子流图如图1~2所示。
2.4 基质效应的考察
本研究通过标准曲线斜率法[14]考察基质效应,基质效应表示为:基质效应(ME%)=(基质匹配标准溶液所作曲线的斜率/无基质标准溶液所作曲线的斜率-1)/100%,基质效应为负值表示存在基质抑制效应,正值表示存在基质增强效应。绝对值在 0~20%之间,表示存在较弱的基质效应;绝对值在 20%~50%之间,表示存在中等的基质效应;绝对值>50%,表示存在较强的基质效应。实验分别用空白基质作溶剂和甲醇作溶剂配制相同浓度的混合系列标准溶液,对30种化合物进行基质效应评价。结果表明:30种化合物均存在不同程度的基质效应,其绝对值为3%~68%,其中二甲双胍基质抑制68%,比格列酮基质抑制65%,格列喹酮基质增强54%;格列比嗪基质抑制62%;其余药物的基质效应均小于25%。综合考虑,本实验采用基质匹配标准溶液的方法消除基质影响,对于检出成分采用加标法定量,对于添加量大于0.1%的成分需采用液相色谱-紫外检测器再次测定以得到准确定量结果。
2.5 线性范围与检出限
采用空白基质提取液作溶剂,选用常见的具有减肥通便效果中药[15]决明子、荷叶、芦荟、大黄、番泻叶、泽泻、首乌和山楂按照等比例混合粉碎,用甲醇稀释100倍超声萃取,过滤待用。配制浓度分别为10,5,1.0,0.5,0.1,0.05,0.01,0.005,0.001 mg/L的系列混合标准溶液,以浓度(X,mg/L)为横坐标,峰面积(Y)为纵坐标,计算回归方程,以信噪比S/N≥3 时的浓度计算检出限,以3倍检出限得到定量下限,结果见表2。每种物质在线性范围内相关系数均大于0.99,检出限为0.003~0.1 mg/kg,能够满足检测要求。
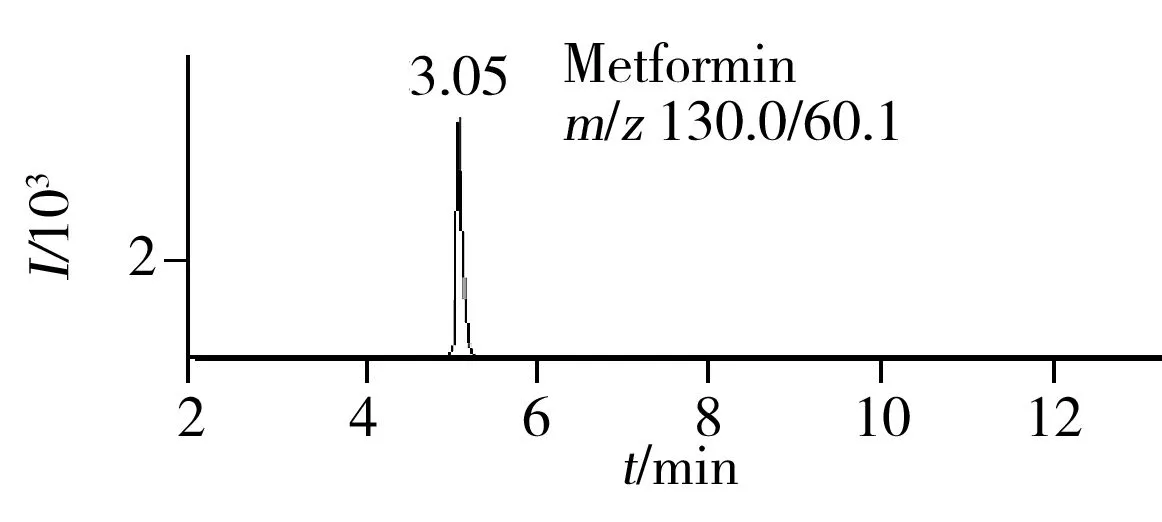







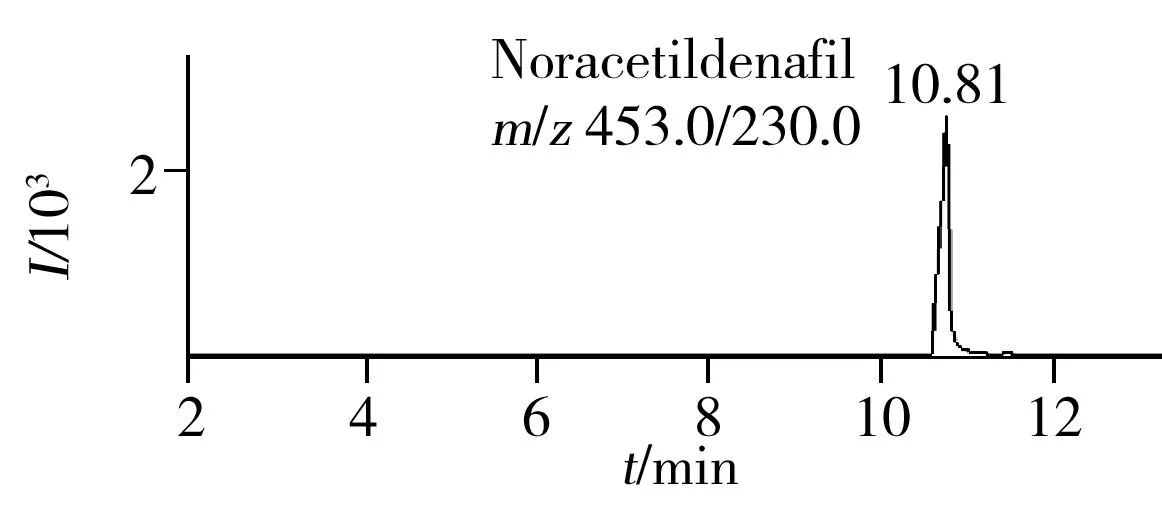

2.6 回收率与精密度
采用经测定不含上述减肥药物的胶囊,添加不同浓度标准溶液,分别进行低、中、高3水平的添加回收率实验,取6次操作的平均值计算回收率及相对标准偏差(RSD),结果见表2。30种减肥化学成分的回收率为70.1%~114.8%,RSD均小于15%。另取1份0.1 mg/L混合标准溶液,在一日内不同时间点(共6个时间点,间隔4 h)测定,得到每种物质的日内精密度均小于6.0%, 表明方法的灵敏度满足定量分析要求。

表2 30种减肥化学药的线性方程、线性范围、相关系数、LOD、LOQ、回收率及精密度(n=6)
*the numbers denoted were the same as that in Table 1
2.7 实际样品的测定
近3年来,本实验室检测了客户委托送检样品300多个,样品类型有胶囊、片剂、原料粉等,其中检出频率最高的为西布曲明和酚酞,少部分检出氯卡色林、氟西汀和二甲双胍成分;另外采用个人购买方式网购了15种不同品牌和来源的样品,结果显示此类非法添加现象仍然存在,部分典型阳性样品的检测结果见表3。

表3 部分阳性样品的测定结果

(续表3)
3 结 论
本研究根据实际检测工作的需要以及多年的积累,建立了一种快速筛查非法添加减肥类化学药的方法,涉及化学药多达30种且具有典型的代表性。该方法覆盖面广、灵敏度高,适用于保健品及中成药非法添加检测,已用于日常检测工作中,为监管相关非法添加提供了有力的技术支持。
[1] Guangxi Quality Supervision Guide Periodical(广西质量监督导报),2013,12:10-11.
[2] Zhang Y F.ChineseBusinessNewspaper(张亦非.中国经营报),2015-08-10C15.
[3] Drug Inspection and Supplementary Inspection Method and Item Number 2006004(药品检验补充检验方法和检验项目批准件编号2006004).
[4] Wang J W,Huang X L,Cao J,Wang G L,Zhang Q S,Ding L X.Chin.J.Chromatogr.(王静文,黄湘鹭,曹进,王钢力,张庆生,丁丽霞.色谱),2014,32(2):151-156.
[5] Li J,Li Y Y,Wen L L.TianjinPharmacy(李静,李倚云,闻琍毓.天津药学),2015,27(2):17-18.
[6] Jiang S Y,Guo C C,Shi F,Gong L P,Zeng S.Chin.J.Pharm.Anal.(姜树银,郭常川,石峰,巩丽萍,曾苏.药物分析杂志),2015,35(8):1447-1452.
[7] Zhu J,Qiu Y J,Shen G F.ChineseTraditionalandHerbalDrugs(朱健,裘一婧,沈国芳.中草药),2014,45(4):509-515.[8] Wang K,Hu Q,Cui Y L,Ji S.Chin.J.Pharm.Anal.(王柯,胡青,崔益泠,季申.药物分析杂志),2008,28(8):1268-1275.
[9] Huang F,Wu H Q,Huang X L,Deng X,Luo H T,Zhu Z X,Lin X S.J.Instrum.Anal.(黄芳,吴惠勤,黄晓兰,邓欣,罗辉泰,朱志鑫,林晓珊.分析测试学报),2013,32(6):699-704.
[10] Shi W F,Gui C,Li X Y,Liu G L.Chin.J.NewDrugs(石卫峰,归成,李晓宇,刘皋林.中国新药杂志),2014,23(2):127-129,133.
[11] The State Food and Drug Administration Asked to Stop the Production of Sibutramine,Sales and Use.Chinese Medicinal Biotechnology(国家药监局要求停止西布曲明的生产、销售和使用.中国医药生物技术),2010,5(6):474.
[12] Zhu F,Ruan L P,Ma Y J,Jie W L,Liu H L.Chin.J.Chromatogr.(朱峰,阮丽萍,马永建,吉文亮,刘华良.色谱),2014,32(1):13-20.
[13] Luo H T,Huang X L,Wu H Q,Zhu Z X,Huang F,Lin X S.J.Instrum.Anal.(罗辉泰,黄晓兰,吴惠勤,朱志鑫,黄芳,林晓珊.分析测试学报),2011,30(12):1329-1337.
[14] He X Q,Xi C X,Tang B B,Wang G M,Peng T,Chen D D,Mu Z D.J.Instrum.Anal.(何小琴,郗存显,唐柏彬,王国民,彭涛,陈冬东,母昭德.分析测试学报),2014,33(6):635-641.
[15] Hou R F,Tao F,Lu H,Shen Y D,Ding X P.Chin.Arch.Tradit.Chin.Med.(侯瑞芳,陶枫,陆灏,沈远东,丁学屏.中华中医药学刊),2015,30(8):1959-1962.
Simultaneous Determination of Thirty Chemical Drugs Illegally Added in Anti-obesity Traditional Chinese Medicine and Health Products by High Performance Liquid Chromatography-Tandem Mass Spectrometry
HUANG Fang*,WU Hui-qin,ZHU Zhi-xin,HUANG Xiao-lan, LUO Hui-tai,LIN Xiao-shan,MA Li-guo,ZHONG Qiao-li,JIANG Ya-lan
(Guangdong Provincial Public Laboratory of Analysis and Testing Technology,China National Analytical Center (Guangzhou),Guangzhou 510070,China )
A high-performance liquid chromatograph-mass spectrometric(LC-MS/MS) method was proposed for the determination of thirty chemical compositions,which were illegally mixed into the antiobesity traditional Chinese medicine and health products.The conditions of extraction and mass parameters were optimized.The drugs were extracted with methonal in ultrasonic condition,separated on a C18chromatographic column using a mixture of acetonitrile and water(containing 10 mmol/L ammonium formate,0.1% formic acid,10%methanol) as mobile phase.The detection of analytes was performed by positive-ion and negative-ion electrospray ionization mass spectrometry(ESI-MS) under multiple reaction monitoring(MRM) mode.The method was proved to be able of covering large area,simple,rapid,accurate and reliable.It was used in screening and examination of illegally added chemicals in Chinese traditional medicine and health food.
antiobesity chemical drugs;liquid chromatography-tandem mass spectrometry(LC-MS/MS);Chinese traditional medicine and health food
10.3969/j.issn.1004-4957.2017.01.001
2016-08-21;
2016-09-20
广东省科技计划项目(2012B040302007)
*通讯作者:黄 芳,硕士,高级工程师,研究方向:分析化学,Tel:020-37656312, E-mail:thymol206@126.com
O657.63;TQ460.72
A
1004-4957(2017)01-0002-07
