分子筛分步催化降解碱木素制备芳香化学品
2017-02-15程毅周自圆李瑞蒋建新
程毅,周自圆,李瑞,蒋建新
(北京林业大学材料科学与技术学院,林业生物质材料与能源教育部工程中心,北京100083)
分子筛分步催化降解碱木素制备芳香化学品
程毅,周自圆,李瑞,蒋建新*
(北京林业大学材料科学与技术学院,林业生物质材料与能源教育部工程中心,北京100083)
本研究采用分步催化的方式对碱木素进行降解,即先利用氢型β分子筛(H-BEA)催化液化碱木素,再采用氢型超稳Y分子筛(H-USY)对液化后的产物进行催化降解。结果表明,相较于非催化条件下的直接降解,分步催化降解将产物中芳香化学品含量从30.6%提升到51.2%。质谱分析(MALDI-TOF)结果表明,降解途径是碱木素中的芳香五聚体被液化为三聚体再进一步降解为芳香二聚体和单体。1H NMR结果表明,第1步催化液化过程破坏了大部分C—O键,而第2步催化降解过程主要以破坏C—C键为主。气相色谱检测到的芳香单体占芳香化学品的16.2%。其中,H-BEA分子筛在液化阶段催化部分酚类烷基化,使得酚羟基的邻位或对位活性降低,防止酚类在第2步催化过程中发生缩合。
碱木素;分步催化降解;芳香化学品;质谱分析(MALDI-TOF);1H NMR
木质素主要是由愈创木基、紫丁香基和对羟苯基等基本结构单元通过C—O键和C—C键结合而成[1-2]。在碱法制浆过程中,木质素会被大量脱除,脱除后的木质素通常会被当做固体燃料直接燃烧,只有少量碱木素被当做工业原料[3]。然而,木质素的芳香基结构决定了这种工业副产品可以作为石油的替代品生产苯和苯酚及其同系物[4]。随着能源问题和环境问题日益严重,工业木质素的高值化利用具有十分重要的意义。
催化降解可以有效地将木质素转化成小分子化学品,具体方式包括催化加氢、催化氧化和催化液化3种方式[5]。其中,催化加氢和催化氧化需要提供额外的氢源或氧源,工业成本相对较高。催化液化可以将木质素在溶液中直接降解成小分子碎片,操作相对简单。然而,由于C—O键或C—C键断裂后会形成反应活性较高的酚羟基或自由基,降解得到的碎片又会重新聚合,很难进一步得到分子量更小的芳香族化合物[6]。因此,防止降解碎片的缩聚是木质素催化液化的关键问题。
酚羟基是木质素降解过程中引发缩合的一个重要官能团。Huang等[7-8]发现,在300℃条件下,乙醇作为溶剂,CuMgAlOx催化剂可以有效地将碱木素降解得到的酚羟基转化为甲氧基,从而保护降解碎片,防止其进一步的缩合。Singh等[9]认为,在非催化条件下,碱木素在甲醇溶液中,200℃就已经可以破坏大部分的C—O键,实现碱木素的初级液化(碎片化)。综合以上报道,本研究采用分步催化的方式对碱木素进行降解,即先在较低温度下破坏碱木素中的C—O键,同时采用氢型β分子筛(H-BEA)对得到的酚羟基进行烷基化,然后再将降解得到的液体采用氢型超稳Y分子筛(H-USY)在较高温度下催化裂化,以期为碱木素高效催化转化为芳香类化学品提供理论和应用技术支撑。
1 材料与方法
1.1 原料制备
试验所采用的黑液取自山东晨鸣造纸集团,为杨木经过氢氧化钠-蒽醌法制浆副产物。为了消除木质素降解过程中碱的影响,本研究采用脱碱木素作为原料。具体操作如下:将黑液稀释10倍,缓慢滴加4 mol/L HCl,充分搅拌,调至pH=2。静置过夜后,离心分离,将得到的固体烘干后水洗,再次离心分离,重复3~5次,直至得到的固体为中性。
研究采用的H-BEA分子筛(Si/Al=25)和H-USY分子筛(Si/Al=14)购自南开催化剂厂。实验中采用的甲醇(分析纯)和对甲苯磺酸(TsOH,分析纯)购自北京化工厂。
1.2 碱木素分步催化降解与产品分离
碱木素两步降解如图1所示,两步反应均在25 mL的水热釜中进行:
第1步,将0.5 g碱木素、0.1 g H-BEA分子筛和10 mL甲醇加入到反应釜中,密封后放入指定温度的烘箱中,反应1 h。反应结束后将反应釜急速冷却至室温,过滤分离釜内混合物。分离后的固体采用甲醇洗至洗出液无色,烘干后称量,计算未反应固体残余质量。

图1 分子筛分步催化碱木素降解流程图Fig. 1 The flow diagram of alkali lignin staged degradation over zeolites catalysts
第2步,将分离后的液体全部转移到另一只25 mL反应釜中,并加入1 mL去离子水和0.25 g H-USY分子筛,密封后放入250℃烘箱中,反应1 h。反应结束后,反应液体直接采用气相色谱测定单体质量,并将所得液体在35℃下真空干燥4 h,除去甲醇和水,称量计算液体得率;固体部分经甲醇洗至无色后烘干,称量计算残余率。
液体烘干后,将其溶解到乙酸乙酯中,其中乙酸乙酯可溶部分为芳香化学品[10],乙酸乙酯不溶部分为低聚体。各组分定量公式如下:
过程1固体残余率=(过程1固体量-催化剂量)/起始木素量
过程2固体残余率=(过程2固体量-催化剂量)/起始木素量
总液体得率=过程2分离后烘干液体量/起始木素量
低聚体得率=烘干液体中乙酸乙酯不溶量/起始木素量
芳香化学品得率=总液体得率-低聚体得率
1.3 原料与降解产品表征
原料与降解产品的1H NMR分析采用Bruker-500 核磁共振谱仪进行检测,将25 mg木素样品溶解于0.5 mL DMSO-d6中,室温下扫描64次。分子量采用基质辅助激光解吸飞行时间检测器(MALDI-TOF)检测,仪器采用AB Sciex TOF/TOF 5800, DHB溶液(溶剂为60% ACN)作为基质。检测时,取1 μL样品点在MALDI板上,扫描范围为100~2 500,激光强度为3 500。气相色谱(GC)采用Aglient 7890B,HP-5MS色谱柱,正十八烷作为内标物,起始温度为50℃,以10℃/min的升温速率升温至250℃,保留时间5 min,GC-MS定性得到的谱图采用NIST库进行检索。
2 结果与分析
2.1 碱木素分步催化降解结果
不同催化条件下碱木素直接降解和分步催化降解的结果见表1。从实验组1到3可以看出,碱木素在250℃直接液化时,固体残余率为17.2%,得到的芳香化学品含量占原木素含量的30.6%。然而,相同条件下加入H-USY或H-BEA后,固体残余量反而增加,得到的芳香化学品含量也相应降低。这与Deepa等[10]得到的结果相反,即催化剂的加入并没有起到积极作用。这种结果的原因为碱木素的来源不同:Deepa等[10]所采用的木素为麦草碱木素,以断裂C—O键为主;而本研究采用的杨木碱木素,其结构与麦草碱木素存在差异,相同反应条件下的结果并不相同。
通过分步催化降解可以显著改变液体产品中芳香化学品的得率。首先液化过程可以使木素以液体的形式与分子筛进行接触,提高反应效率。从实验组5中可以看出,非催化条件下得到的液化油被分子筛进一步降解,芳香化学品含量从直接反应的30.6%升高到45.7%,虽然总的固体残余含量也明显升高,但是得到的液体中芳香化学品的含量占了绝大部分比例,这对于后期分离也是有益的。需要注意的是,在150℃条件下直接液化得到的芳香化学品含量没有明显变化。这是因为该温度下碱木素中的C—O键并不能被破坏,而固体残余中主要是分子量较大、不能被甲醇溶解的木素[11-12]。这一部分木素的排除,使得后期反应过程得到的芳香化学品含量明显降低。
催化剂的加入可以提高芳香化学品的得率,这是因为H-BEA可以催化苯酚进行烷基化反应。其中,对于酚羟基的O-烷基化,得到的芳香醚类产品可以降低邻位和对位的反应活性;对于酚羟基的C-烷基化,邻位或对位被填补,不会再和羰基类物质发生缩合反应,使得液化后的产品可以进一步降解得到小分子的芳香化学品[13]。对甲苯磺酸是木材液化常用的催化剂,在150℃就可以液化大部分原料。在本研究中,对甲苯磺酸也表现出较好的液化能力,固体残余量仅有13%。然而,最终的芳香化学品含量并不高,这可能是因为酸性催化剂主要以攻击Cα位为主,形成的正电荷很容易与亲电基团进行反应,使得到的小分子重新缩合,不利于碱木素的降解[14]。因此,从以上结果中可以发现,将碱木素液化并进行烷基化保护,可以有效地提高降解效率,获得更多的芳香化学品。

表1 分步催化碱木素降解产物得率
注:a.过程1反应溶剂为甲醇,反应时间1 h;b.过程2反应溶剂为甲醇和水(体积比10∶1),反应时间1 h。
2.2 MALDI-TOF分子量分析
基质辅助激光解吸飞行时间检测器(MALDI-TOF)检测到的碱木素、H-BEA分子筛在200℃液化后的木素和H-USY在250℃进一步降解后的木素其分子量变化见图2。从图1a可以看出,碱木素的分子量分布范围为100~800,其中m/z=715,579,421和393的强度要明显高于液化后木素(b)和降解后木素(c)。以木素C9单元为单体计算,质量范围在350~750的木素大概由3~5个C9单元组成[2]。H-BEA催化液化木素,除了m/z=349,其他的峰都明显降低。这说明在200℃,H-BEA催化条件下,可以有效降解一部分大分子木素。当木素液化后,得到的液体在250℃条件下采用H-USY催化,m/z大于300的分子都明显降解。MALDI-TOF结果说明,分步催化降解碱木素的基本降解途径是碱木素中的五聚体液化为三聚体再进一步催化转化为二聚体或单体。

图2 碱木素(a)、H-BEA催化液化木素(b)和H-USY催化降解木素(c)的分子量变化Fig. 2 The molecular weight change of alkali lignin, liquefied lignin and degraded lignin
2.31H NMR分析
碱木素、H-BEA分子筛在200℃液化后木素和H-USY在250℃催化后木素的1H NMR谱图和积分结果见图3、表2。结果归属参考文献[15-16],对比积分结果可以发现,液化后δ0~2.25所对应的脂肪族烷烃含量增加,说明液化过程断裂了部分C—O键产生了甲基或亚甲基基团。相对应的,δ4.05~6.00处的脂肪族C—O键大量减少,一方面可能是由于碱木素中残留的糖被降解,另一方面是由于脂肪族C—O键的断裂。δ3.45~4.05处对应的甲氧基含量有少量增加,说明在H-BEA的催化作用下,一部分的酚羟基转化成了甲氧基,但是增长幅度不大,说明了该条件下只有少部分的酚羟基发生了烷基化反应。相应的,δ8.00~9.35处酚羟基的减少也证明了这一点。值得注意的是,芳香环上的H含量也有所减少,这可能是因为液化过程使得总的官能团数量增加,造成了芳香环相对比例降低。
对比H-USY催化后的木素和液化木素发现二者1H NMR 结果的总体变化幅度较小,δ0.00~2.25处代表的脂肪族烃类含量有少量降低,可能由于部分高键能的连接键在第2步催化阶段被破坏,而键能较低的C—O连接键在液化阶段已经被破坏。脂肪烃类含量的减少,说明有一部分C—C键的断裂,同时也说明在该体系中,C—C键的断裂是第2步催化过程得到芳香单体的主要方式。

图3 碱木素、H-BEA催化液化木素和H-USY催化降解木素的1H NMR 谱图Fig. 3 1H NMR spectrum of alkali lignin, liquefied lignin and degraded lignin
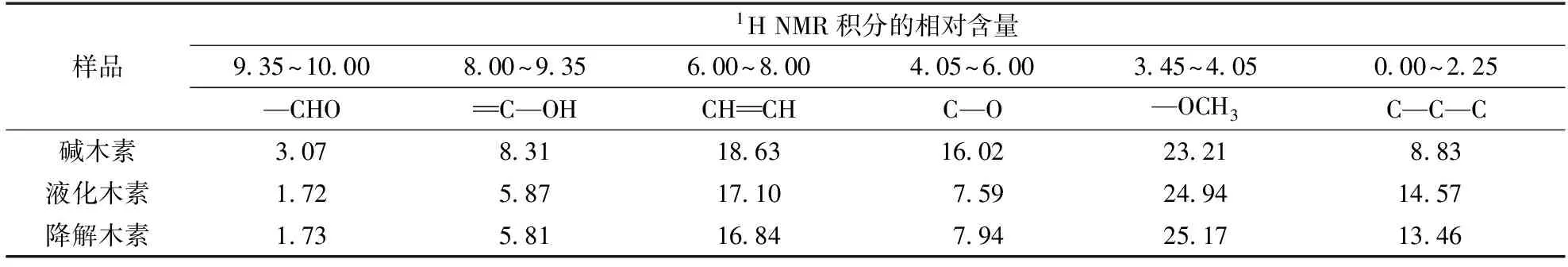
表2 碱木素、H-BEA催化液化木素和H-USY催化降解木素的1H NMR 积分结果
2.4 GC-MS分析
最终产品中主要芳香单体的含量见表3,其中以酚类产品为主,包括少量的芳香醚类化合物,这可能是由液化阶段酚羟基甲基化得到的产品[13]。而整体产品中,S型酚的种类和含量均高于G型酚,一方面是由于杨木结果中S型结构含量高于麦草[17],另一方面可能是酚羟基邻位容易与其他带电基团缩合[18-20],形成低聚体而难以被气相色谱检测到。
通过分步催化,最终得到的芳香化学品含量(质量分数)为51.2%,其中芳香单体占芳香化学品含量的16.2%(GC检测)。相比较,Deepa等[10]采用麦草碱木素为原料,在250℃条件下采用H-USY直接降解,得到60%的芳香化学品。这说明相同催化体系,对于不同碱木素原料,需要选择适当催化剂,以提高芳香化学品的得率[21-22]。

表3 分步催化降解主要芳香单体含量
3 结 论
本研究采用分步催化降解的方式对杨木碱木素进行降解,即先将碱木素在200℃、H-BEA分子筛催化条件下液化,再将液化后的产品在250℃、H-USY催化条件下进行第2步催化,结论如下:
1)相较于非催化条件下的直接降解,分步催化降解将芳香化学品含量(质量分数)从30.6%提升到51.2%,H-BEA在液化阶段烷基化了部分酚羟基,使得在接下来的催化过程中芳香化学品含量有所提升。
2)MALDI-TOF结果说明了分步催化的可能降解途径是碱木素中的芳香五聚体液化为芳香三聚体再进一步催化为芳香二聚体和单体。
3)1H NMR 结果表明,液化过程可以破坏大部分C—O键的连接,第2步催化过程主要以破坏C—C键为主;通过气相色谱可以检测到芳香化学品中含有16.2%的芳香单体。
[1]郭思勤, 游婷婷, 周天,等. 酸催化离子液体预处理芦竹酶木质素结构研究[J]. 林业工程学报, 2016, 1(2):82-87. GUO S Q, YOU T T, ZHOU T, et al. Structure elucidation of cellulolytic enzyme lignin fromArundodonaxLinn. after acid-catalyzed ionic liquid pretreatment[J]. Journal of Forest Engineering, 2016, 1(2):82-87.
[2]VANHOLME R, DEMEDTS B, MORREEL K, et al. Lignin biosynthesis and structure[J]. Plant Physiology, 2010, 153(3):895-905.
[3]LORA J H, GLASSER W G. Recent industrial applications of lignin: asustainable alternative to nonrenewable materials[J]. Journal of Polymers & the Environment, 2002, 10(1):39-48.
[4]ZAKZESK J, BRUIJNINCX P C A, JONGERIUS A L, et al. The catalytic valorization of lignin for the production of renewable chemicals[J]. Chemical Reviews, 2010, 110(6):3552-3599.
[5]LI C Z,ZHAO X C,WANG A Q,et al. Catalytic transformation of lignin for the production of chemicals and fuels[J]. Chemical Reviews, 2015, 115(21):11559-11624.
[6]SONG Q, WANG F, CAI J, et al. Lignin depolymerization (LDP) in alcohol over nickel-based catalysts via a fragmentation-hydrogenolysis process[J]. Energy & Environmental Science, 2013, 6(3):994-1007.
[7]HUANG X M, KORNYI T I, BOOT M D, et al. Catalytic depolymerization of lignin in supercritical ethanol[J]. Chemsuschem, 2014, 7(8):2276-2288.
[8]HUANG X M, KORNYI T I, BOOT M D, et al. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics[J]. Green Chemistry, 2015, 17 (11):4941-4950.
[9]SINGH R, PRAKASH A, DHIMAN S K, et al. Hydrothermal conversion of lignin to substituted phenols and aromatic ethers[J]. Bioresource Technology, 2014, 165(8):319-322.
[10]DEEPA A K, DHEPE P L. Lignin depolymerization into aromatic monomers over solid acid catalysts[J]. ACS Catalysis, 2015, 5(1):365-379.
[11]LIN L Z, YOSHIOKA M, YAO Y G, et al. Liquefaction mechanism of lignin in the presence of phenol at elevated temperature without catalysts. Studies onβ-O-4 lignin modelcompound. II. reaction pathway[J]. Holzforschung, 1997, 51(4):325-332.
[12]SAITO T, PERKINS J H, VAUTARD F, et al. Methanol fractionation of softwood kraft lignin:impact on the lignin properties[J]. Chemsuschem, 2014, 7(1):221-228.
[13]SAD M E, PADRC L, APESTEGUA C R. Study of the phenol methylation mechanism on zeolites HBEA, HZSM5 and HMCM22[J]. Journal of Molecular Catalysis A: Chemical, 2010, 327(1/2):63-72.
[14]JASIUKAITYTGROJZDEK E, KUNAVER M, CRESTINI C. Lignin structural changes during liquefaction in acidified ethylene glycol[J]. Journal of Wood Chemistry & Technology, 2012, 32(4):342-360.
[15]NAGY M, KOSA M, THELIANDER H, et al. Characterization of CO2precipitated kraft lignin to promote its utilization[J]. Green Chemistry, 2010, 12(1):31-34.
[16]PRINSEN P, RENCORET J, GUTIRREZ A, et al. Modification of the lignin structure during alkaline delignification of eucalyptus wood by kraft, soda-AQ, and soda-O2cooking[J]. Industrial & Engineering Chemistry Research, 2013, 52(45):15702-15712.
[17]WU M, ZHAO D H, PANG J H, et al. Separation and characterization of lignin obtained by catalytic hydrothermal pretreatment of cotton stalk[J]. Industrial Crops & Products, 2015, 66(66):123-130.
[18]BARBIER J, CHARON N, DUPASSIEUX N, et al. Hydrothermal conversion of lignin compounds. A detailed study of fragmentation and condensation reaction pathways[J]. Biomass and Bioenergy, 2012, 46:479-491.
[19]LIN L, YAO Y, SHIRAISHI N. Liquefaction mechanism ofβ-O-4 lignin model compound in the presence of phenol under acid catalysis. Part 1. Identification of the reaction products[J]. Holzforschung, 2001, 55(6): 617-624.
[20]LIN L, NAKAGAME S, YAO Y, et al. Liquefaction mechanism ofβ-O-4 lignin model compound in the presence of phenol under acid catalysis. Part 2. Reaction behaviour and pathways[J]. Holzforschung, 2001, 55(6):625-630.
[21]TOLEDANO A, SERRANO L, LABIDI J. Improving base catalyzed lignin depolymerization by avoiding lignin repolymerization[J]. Fuel, 2014, 116:617-624.
[22]NAGY M, KOSA M, THELIANDER H, et al. Characterization of CO2precipitated kraft lignin to promote its utilization[J]. Green Chemistry, 2010, 12(1):31-34.
Producing aromatic chemicals from stepwise degradation ofalkali lignin over zeolites catalysts
CHENG Yi, ZHOU Ziyuan, LI Rui, JIANG Jianxin*
(CollegeofMaterialScienceandTechnology,MOEEngineeringResearchCenterofForestryBiomassMaterialsandBioenergy,BeijingForestryUniversity,Beijing100083,China)
Lignin is the second most abundant natural polymer and consists of methoxylatedphenylpropane units cross-linked by C—C and C—O bonds. This renewable resource is of interest as an alternative to petroleum for the production of valuable chemicals like phenols, benzene, and toluene. In the present study, aromatic chemicals were produced from stepwise degradation of alkali lignin over zeolites. Namely, the lignin was first liquefied over H-BEA zeolite, thus the obtained oil was catalytic refined over H-USY zeolite. The results showed that the yield of aromatic chemicals increased from 30.6% to 51.2% compared to single step reaction. In the process, parts of the phenolic hydroxyl groups were transformed into aromatic ethers over H-BEA at first stage and the C—C linkage were broken over H-USY at second stage. According to molecular weight analysis results by matrix assisted laser desorption/ionization time of flight (MALDI-TOF), it could be concluded that lignin first degradated to 4 or 5 C9 units followed by further degradation to aromatic trimmers, and the aromatic trimmer then degraded to aromatic dimmer or monomers.1H NMR demonstrated that the C—O linkages were broken during the liquefaction stage, and the refining stage only broke some C—C linkage. Finally, 16.2% monomers of aromatic chemicals were detected by gas chromatography. A possible explanation for the improved output of aromatic species over stepwise degradation is that the alkylation of phenolic hydroxyl groups can facilitate lignin depolymerization; the resulted aromatic ethers are less active toreact with lignin in the second catalytic degradation stage.
alkali lignin; stage degradation; aromatic chemicals; MALDI-TOF;1H NMR
2016-07-03
2016-10-18
国家自然科学基金项目(31070510);科技部国际科技合作专项(2014DFG32550)。
程毅,男,博士生,研究方向为生物质催化转化。通信作者:蒋建新,男,教授。E-mail:jiangjx@bjfu.edu.cn
TQ35
A
2096-1359(2017)01-0057-06
