散发性低钾性周期性麻痹KCNJ18 KCNJ12基因分析
2017-01-17梅志忠黄晓芸付文金方浩威黄益洪余映丽陈建军王明霞官少兵
梅志忠 林 菡 黄晓芸 付文金 方浩威 黄益洪 余映丽 陈建军 王明霞 官少兵
广东东莞市厚街医院神经内科 东莞 523945
散发性低钾性周期性麻痹KCNJ18 KCNJ12基因分析
梅志忠 林 菡 黄晓芸 付文金 方浩威 黄益洪 余映丽 陈建军 王明霞 官少兵
广东东莞市厚街医院神经内科 东莞 523945
目的 通过对散发性低钾性周期性麻痹KCNJ12、KCNJ18基因测序分析,探讨基因突变的相关性。方法 取我院住院的52例散发性低钾性周期性麻痹患者和10例健康对照者血样,应用PCR和测序技术,进行KCNJ12、KCNJ18基因编码区PCR、测序比对。结果 通过对52例患者和10例健康对照者KCNJ12、KCNJ18基因扩增测序比对,均未发现明显异常。结论 未发现散发性低钾性周期性麻痹患者KCNJ12、KCNJ18基因编码区序列存在突变。
散发性低钾性周期性麻痹;KCNJ18;KCNJ12;基因;突变
低钾性周期性麻痹是一组原发性伴有低钾血症的突发性肌肉迟缓性瘫痪肌病。常因剧烈运动后大汗、饱餐、寒冷刺激及应用胰岛素、糖皮质激素后诱发。HypoKPP可分为家族性和非家族性。家族性低钾性周期性麻痹(FPP)是一常染色体显性遗传性疾病,是由于编码骨骼肌特异性电压门性Na+通道Nav1.4(SCN4A)或Ca2+通道Cav1.1(CACNA1S)蛋白基因突变所致[1-3]。约60%的家族性低钾性周期性麻痹是由CACNA1S突变所致,被称为hypoPP-1型,约20%的患者是由SCN4A基因突变导致的,被称为hypoPP-2型,大约3.5%的患者是由KCNJ18基因突变导致。
非家族性低钾性周期性麻痹(SPP)包括甲亢性和散发性低钾性周期性麻痹[4]。甲亢性低钾性周期性麻痹(HPP)以甲亢患者继发低钾性周期性麻痹为特点,其临床表现与家族性周期性麻痹相类似。亚洲人种男性和拉丁美洲男性人的非家族性低钾性周期性麻痹发病率明显高于高加索人种[5]。在亚洲人种和拉丁美洲人种的甲亢男性患者中,其发生低钾性周期性麻痹的发病率高达10%,而高加索人种的发病率还不到0.1%[6]。
目前,散发性低钾性周期性麻痹(SPP)的发病机制尚不清楚。散发性低钾性周期性麻痹其临床表现与家族性低钾性周期性麻痹类似,但不具有家族性。一般认为与家族性低钾性周期性麻痹具有相似的发病机制,可能是由于离子通道病变所引起,值得深思的是只有极少数散发性周期性麻痹患者检测到SCN4A和CACNA1S基因突变[7]。
KCNJ18基因编码内向整流性钾离子通道蛋白,有研究认为可能与甲亢性低钾性周期性麻痹和散发性低钾性周期性麻痹有关,但也只有极少数散发性周期性麻痹患者可以检测到KCNJ18基因突变[8]。KCNJ12是KCNJ18的同源基因,同样编码钾离子通道蛋白,其DNA及RNA序列与KCNJ18高度相似。虽有动物实验研究KCNJ12基因与低钾性周期性麻痹的关系,但还没有人类低钾性周期性麻痹的基因研究报道。鉴于KCNJ18和KCNJ12的序列和功能高度同源性,有必要同时研究两者对周期性麻痹的影响,明确KCNJ12的突变是否影响周期性麻痹。
本研究就住院的散发性低钾性周期性麻痹中国患者进行了KCNJ18、KCNJ12基因进行测序比对分析。
1 对象与方法
1.1 研究对象 52例均为我院2014—2015年收治的散发性低钾性周期性麻痹患者,10健康对照者均告知知情同意书并签字。均符合散发性低钾性周期性麻痹的诊断标准:表现为发作性低钾性迟缓性瘫痪,血钾低于2.5 mmol/L,无家族史;排除胃肠道钾丢失、TTKG>3、甲亢及药物性低钾等。患者均为男性,平均年龄(28±5)岁;10例健康对照组平均年龄(29±4)岁,均为男性,与患者组相比年龄无显著性差异。
1.2 方法
1.2.1 DNA 的提取:采集每个研究对象外周血3 mL,按照1:3加入9 mL RNALock血液RNA稳定剂(厂家:TIANGEN,货号:DP440-01),颠倒混匀,从中吸取1 mL混合液,采用血液基因组DNA提取试剂盒(厂家:TIANGEN,货号:DP348)提取基因组DNA。剩余混合液保存于-20℃,用于后续RNA提取。
1.2.2 序列比对及引物设计:利用NCBI数据库在线BLAST工具(http://blast.ncbi.nlm.nih.gov/Blast.cgi)比对人类KCNJ18和KCNJ12基因组序列差异。根据序列比对结果,Primer premier 5.0软件设计引物,用于研究对象扩增KCNJ12和KCNJ18的基因组序列。
1.2.3 PCR扩增:PCR反应体系25 μL,含DNA 50 ng,引物0.12 μLmol /L,dNTPs 200 μLmol/ L,TaqDNA 聚合酶1 U(日本,TaKaRa 公司)。PCR 反应条件:在Cetus2400 热循环仪(美国,PE 公司),预变性94 ℃ 5 min;变性94 ℃ 40 s,退火温度由具体引物决定,延伸72 ℃ 60 s,共35循环;延伸7 min。应用2%琼脂糖电泳检测PCR产物。
1.2.4 测序及序列分析:PCR产物送至北京六合华大基因科技股份有限公司,采用Sanger法进行双向测序。序列比对和突变位点鉴定采用突变分析软件Mutation Surveyor。
2 结果
2.1KCNJ18和KCNJ12序列结构分析 已公布的人类KCNJ18 DNA序列(ACCESSION:NG_033093)全长12090bp,位于第17染色体,由3个外显子和2个内含子组成,mRNA(ACCESSION:NM_001194958.2)的剪接位置为:1~192,3 461~3 582,10209-12090,长度2 196 bp,其中编码区长度1302bp,位于10265-11566。此外,最近的序列预测发现KCNJ18存在一个转录变异体(transcript variant)X1(ACCESSION:XM_005276919.2),长度3 381 bp,其中编码区长度1 608 bp。
人类KCNJ12 DNA序列(ACCESSION:NG_042809)全长43 481 bp,位于第17染色体,同样由3个外显子和2个内含子组成,mRNA(ACCESSION:NM_021012.4)的剪接位置为:1~527、32 133~32 254、38 901~43 481,长度5 230 bp,其中编码区长度 1302 bp,位于38957~40258。此外,最近的序列预测发现KCNJ12存在二个转录变异体X1(ACCESSION:XM_005256625.3)和X2(ACCESSION:XM_011523831.1),X1长度4931 bp,其中编码区长度1 302 bp,X2长度4 819 bp,其中编码区长度1 302bp。
序列比对发现,KCNJ18编码区序列与KCNJ12的相似度高达1 290/1 302(99.08%),仅12个碱基有差异,未出现序列空位(Gaps)。KCNJ18基因前导区(编码区上游1 0264 bp)与KCNJ12的序列相似度高达10 027/10 300(97.35%),Gaps占129/10 300(1.25%)。KCNJ18尾部区(编码区下游507 bp)与KCNJ12的序列相似度高达97%(509/527),Gaps占0.76%(4/527)。见图1。
mRNA序列比对发现,KCNJ18的mRNA与KCNJ12的三个转录本序列的同源百分百均为91%,序列相似度为98%。KCNJ18的转录变异体X1与KCNJ12的三个转录本序列的同源百分百均为87%,序列相似度为98%。见图2。

图1 KCNJ18 mRNA与KCNJ12三个转录本序列比对
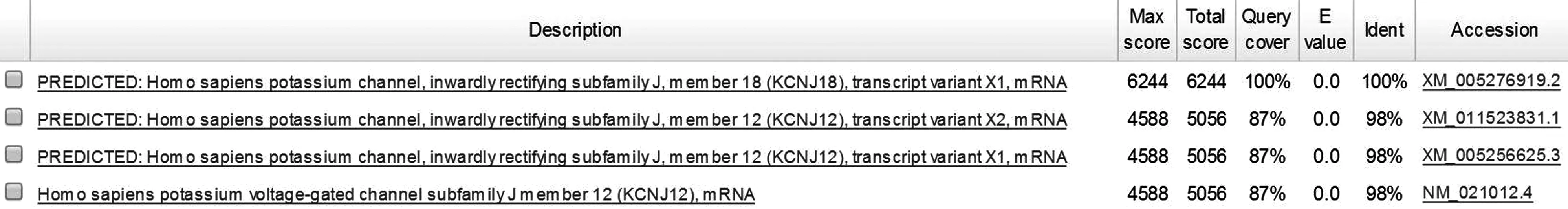
图2 KCNJ18的转录变异体X1与KCNJ12三个转录本序列比对
2.2 测序片段引物设计 由于KCNJ18和KCNJ12基因组序列太长,本研究只针对其编码区进行设计PCR引物。鉴于KCNJ18和KCNJ12编码区及其侧翼序列高度相似,且缺少序列空位,本研究在编码区及其侧翼序列(各100bp)共1502 bp范围内设计3对公共引物(KCNJ-1F:CACCAGCCCAGCCAGACA,KCNJ-1R:CTGTGCCCGCTTCTTGGG,KCNJ-2F:TGCATCATCGACTCCTTCATG,KCNJ-2R:GAGGGCACCTCATAGGTCTTG,KCNJ-3F:CACAGCCATGACCACCCA,KCNJ-3R:CCCGTTCTGCTCAAACCC),其扩增片段长度分别为670bp、550bp和475bp,扩增范围覆盖两个基因的编码区。
2.3 测序片段序列比对 将3对公共引物在52例研究对象和10例健康对照者基因组DNA中的PCR产物进行测序,结果表明,3对引物均可同时在KCNJ18和KCNJ12基因上扩增出目的片段。由于KCNJ18和KCNJ12的编码区序列高度同源,不存在序列空位,从PCR产物测序峰图可分辨出KCNJ18和KCNJ12序列(图3)。利用突变分析软件Mutation Surveyor比对52例研究对象在KCNJ18和KCNJ12编码区序列上的差异,结果未发现任何差异。
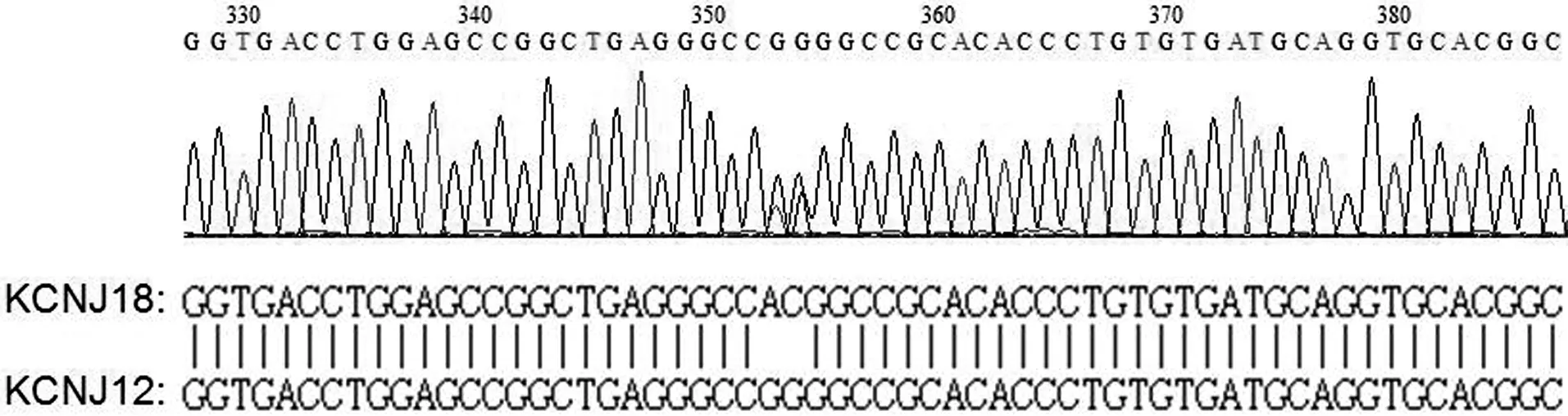
图3 引物KCNJ-1的PCR产物测序峰图
3 讨论
SPP是亚洲人引起低钾型周期性麻痹的第二大主要原因,其发病明显高于高加索人种。因此,以大陆华人作为研究对象对揭示SPP的发病机制具有典型的代表性,具有重要意义。由于散发性低钾性周期性麻痹与家族性低钾性周期性麻痹具有类似的临床表现特征特征,所以一直被认为其发病机制可能与家族性低钾性周期性麻痹同样是由常染色体显性遗传缺陷导致,或者是不完全显性的基因突变,对父母影响轻微,也有可能基因突变没有影响到父母,尽管SCN4A和CACNA1S基因研究不能得到证实。有报道认为KCNJ18基因突变引起散发性低钾性周期性麻痹,但可惜的是只有极少数患者可以检测得到基因的病态突变[9]。KCNJ12目前尚没有人类相关低钾性周期性麻痹的报道。
尽管SPP的临床表现与FPP非常相似,但其临床症状比较轻微,发病年龄晚、发作频率低、缺乏明显诱发因素等特点,只有少数SPP患者发现有CACNA1S或SCN4A基因突变。因此,Sung CC认为,可能与FPP有所不同,代表着hypoKPP独特的亚型[4]。
本研究表明,大陆华人散发性低钾性周期性麻痹患者DNA未发现致病性KCNJ18和KCNJ12基因DNA突变。在本研究的52例患者中,通过KCNJ18、KCNJ12基因扩增测序,与健康人群比较,均未发现有意义的基因突变。回顾相关研究报告,显示不论是华人还是高加索人患者,尽管有点突变报道,但发现率极低,难以解释SPP的发病机制。
内向性钾离子整流通道是肌肉关键性部分,决定着肌肉的静息电位、电兴奋性调节、动作电位去极化、钾离子从T管系统中清除,Kir2通道位于细胞膜和T-管,Kri2的丰度对肌肉的功能至关重要。钾离子通道通过与其他Kir亚单位重装配影响影响肌肉的特性。内向性整流钾通道由亚单位的同源四聚体或异源四聚体装配形成。Kri2.1、Kri2.2、Kri2.3亚单位能重装配形成异源四聚体。Kir2.6在细胞表面很少表达,主要存在内质网,但与其他Kir2.x很容易形成再装配异源四聚体,通过内质网的显性负性保留对内向性整流通道调控起到重要的调节作用。因此认为Kir2.6是一个存在于内质网的调节亚基。Kir2.6和Kir2.1或Kir2.2的联合促进了异源四聚体在内质网的保留,内向性整流通道在细胞膜上的数量取决于Kir2.1、Kir2.2和Kir2.6亚单位之间的比例。我们推测,要保持健康个体内环境的稳定,就要使细胞表面通道有一个稳定的表达数量。认为内质网保留是控制肌肉兴奋性重要机制[10]。但在人类细胞系(HEK细胞)培养中发现Kir2.6通道是在细胞表面表达,Kir2.6和Kir2.1共表达产生Kir2.6和Kir2.1通道亚单位复合功能特性电流,Kir2.6在细胞表面表达产生细胞表面电流。KCNJ18基因如何导致细胞电生理改变还不清楚,需要进一步研究。但不论kir2.6是在细胞表面表达还是存在于内质网,mRNA低表达都可能影响其功能。
由于KCNJ18和KCNJ12编码区及其侧翼序列高度同源,其PCR引物必须根据两者序列的同源性进行设计,否则容易因为引物与目标序列错配导致扩增产物测序失败,或者测序结果出现假阳性。此外,相关研究表明KCNJ18突变与家族性周期性麻痹有关,鉴于KCNJ12与KCNJ18高度同源,而有关KCNJ12是否参与家族性周期性麻痹尚未见相关报道,有必要对其进行研究。
尽管目前尚未在KCNJ18和KCNJ12的编码区发现序列突变,但这两个基因的编码区只占其DNA序列的一小部分,大部分序列位于前导区和尾部区,目前未能排除这两个区域存在突变。非编码区的突变可影响基因的表达,从而影响基因的功能。目前的序列预测表明KCNJ18和KCNJ12存在5个转录本,说明两者的基因表达比想象更为复杂。因此,有必要研究这两个基因在散发性低钾性周期性麻痹患者与健康对照在转录水平上的差异。
综上所述,本研究表明,散发性低钾性周期性麻痹在内向性整流钾离子通道基因KCNJ18/KCNJ12基因编码区序列测序比对未发现与健康人群有差异。由于基因表达过程的复杂性,还需对其进一步研究,除寻找基因突变性致病的可能外,也不排除基因表达调控异常的可能性。
真诚感谢:广东省农业科技研究所红颜彬博士在课题实验研究的大力支持与指导。
[1] Ptacek LJ,Tawil R,Griggs RC,et al.Dihydropyridine receptor mutations cause hypokalemic periodic paralysis[J].Cell,1994,77(6):863-868.
[2] Venance SL,Cannon SC,Fialho D,et al.The primary periodic paralyses:diagnosis,pathogenesis and treatment[J].Brain,2006,129(pt 1):8-17.
[3] Sternberg D,Maisonobe T,Jurkat-Rott K,et al.Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A[J].Brain,2001,124(6):1 091-1 099.
[4] Sung CC,Cheng CJ,Lo YF,et al.Genotype and phenotype analysis of patients withsporadic periodic paralysis[J].Am J Med Sci,2012,343(4):281-285.
[5] Kung AW.Clinical review:thyrotoxic periodic paralysis:a diagnostic challenge[J].J Clin.Endocrinol Metab,2006,91(7):2 490-2 495.
[6] Kelley DE,Gharib H,Kennedy FP,et al.Thyrotoxic periodic paralysis.Report of10 cases and review of electromyographic findings[J].Arch Intern Med,1989,149(11):2 597-2 600.
[7] Lin SH,Hsu YD,Cheng NL,et al.Skeletal muscle dihydropyridi ne-sensitivecalcium chann el(CACNA1S)gene mutations in Chinese patients with hypok periodic paralysis[J].Am J Med Sci,2005,329(2):66-70.
[8] Ryan DP,da Silva MR,Soong TW,et al.Mutations in potassium channelKir2.6cause susceptibility to thyrotoxic hypokalemic periodic paralysis[J].Cell,2010,140(1):88-98.
[9] Kuhn M,Jurkat-Rott K,Lehmann-Horn F.RareKCNJ18 variants do not explain hypokalaemicperiodic paralysis in 263 unrelated patients[J].J Neurol Neurosurg Psychiatry,2016,87(1):49-52.
[10] Dassau L,Conti LR,Radeke CM,et al.Kir2.6 Regulates the Surface Expression of Kir2.x InwardRectifier Potassium Channels[J].J Biol Chem,2011,286(11):9 526-9 541.
(收稿2016-07-12 修回2016-09-15)
广东省科技计划项目,编号:201303B021800031
R 746.3
A
1673-5110(2016)24-0069-03
