磷酸肌酸钠的合成
2017-01-16陈宁
陈 宁
(海南中化联合制药工业股份有限公司,海南 海口 570216)
磷酸肌酸钠的合成
陈 宁
(海南中化联合制药工业股份有限公司,海南 海口 570216)
通过对磷酸肌酸钠的合成研究,确定了磷酸肌酸钠的合成路线,并可以用于工业化生产。该工艺条件与其他路线比较,具有起始原料易得、反应条件温和、产品收率高、操作简便等优点。即以肌酸酐为起始原料与磷酸三乙酯混合,在超声条件下滴加三氯氧磷,缩合反应后得到肌酐磷酰氯,然后用氢氧化钠水解开环得到磷酸肌酸钠粗品,粗品用大孔树脂搅拌吸附和重结晶后得到磷酸肌酸钠精制品,结构经1H-NMR确认,总收率65%,含量大于99.9%,产品质量符合2015版药典要求。
磷酸肌酸钠;肌酸酐;合成
磷酸肌酸是人体内自有的活性物质,是人体重要的能量供应源,为ATP补充能量,而ATP是任何细胞代谢过程中最主要的能量源。在体内CP的生物合成从肾脏开始,由甘氨酸与精氨酸反应形成胍基乙酸,然后在肝脏经甲基化形成肌酸,最后在各组织中被磷酸化后形成CP[3]。
CP于1927年由Eggleton于哺乳动物肌肉中分离得到,经过了50多年的深入研究后,人们才了解到这一药物的许多生理功能,俄国、意大利、英国及瑞典的心脏内、外科中心对该药进行了多次研究及临床应用,我国在心脏手术中使用外源性CP也取得了良好的教果。随着治疗水平和生活水平的不断提高,代谢性心肌保护治疗成为了最前沿的治疗[1]。
自该药上市以来,在临床心脏手术时加入心脏停搏液中保护心肌,缺血状态下的心肌代谢异常等方面,表现出良好的临床疗效。
1 合成方法综述
经文献检索,磷酸肌酸钠主要合成工艺方法如下:
(1)生物酶催化法
文献[2-3]中公开了一种采用从兔肌肉中提取的磷酸肌酸激酶,以磷酸和肌酸为原料,生物酶催化合成磷酸肌酸钠的方法。
实际上,该酶促反应方法没有解决产品在水反应体系中生成后如何分离纯化出来的问题。反而由于动物来源的酶的加入,给静脉注射的磷酸肌酸钠带来潜在的病毒和微生物污染的危险。
(2)化学合成法
方法一:文献[4]用肌酸和三氯氧磷在低温条件下于碱溶液中进行反应,反应完毕除去生成的磷酸钠及未反应的肌酸,滤液中加入氯化钙使之生成磷酸肌酸钙,重结晶后得纯品。此方法纯度不高,钙盐的水溶性不理想,不适于药用。
方法二:文献[5]以肌酸、氧氯化磷与氢氧化钠在低温下反应,以乙醇沉淀并过滤除去部分无机盐,然后加入溴化钡,所得磷酸肌酸钡盐沉淀分离,再用硫酸钠溶液沉淀钡离子,滤液中加入乙醇沉淀得磷酸肌酸钠。该方法步骤较多,工艺条件相对苛刻,且工艺中使用了钡盐,如带进制剂中较危险。也有文献报道对该法进行了研究,但也存在收率不高,产品在那个钡盐含量未能有效控制的问题。
方法三:文献[6]以肌酐为原料,与二苯氧基磷酰氯缩合得二苯氧基磷酰肌酐,再用二氧化铂催化氢解脱去苯基得到磷酸肌酐二钠,最后碱性水解得磷酸肌酸钠。如图1所示。
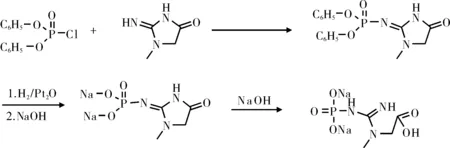
图1 磷酸肌酸钠的合成法1
此方法需要催化氢解,操作繁琐,不适合工业放大生产。
方法四:文献[7]用肌酐和三氯氧磷在回流条件(108 ℃)缩合,得到肌酐磷酰氯,在经过水解开环反应得到磷酸肌酸钠粗品,经过树脂纯化得到磷酸肌酸钠产品如图2所示。
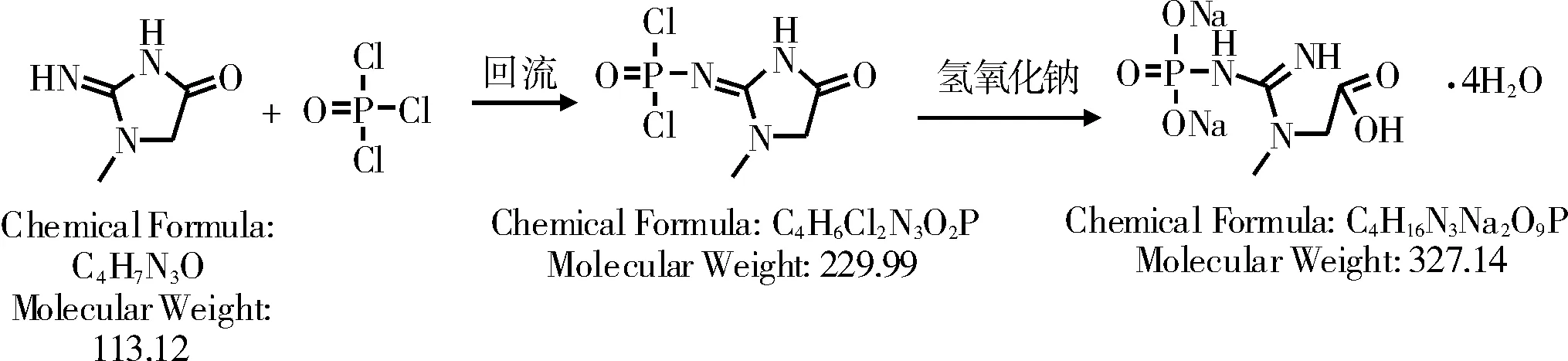
图2 磷酸肌酸钠的合成法2
此方法中,三氯氧磷可蒸馏回收套用;中间体可控,产品纯度高,没有重金属残留污染。只是用交换树脂纯化,操作繁琐,并且反应温度高,不可控,不适合在工业化生产中使用。
2 工艺路线的选择
综合文献报道,我们的工艺路线采用方法四的合成方法,以肌酐为起始原料合成磷酸肌酸钠。与其他路线相比,该路线具有各步收率高,中间体可控,反应条件适合工业化生产,起始原料易得等优点。同时避免使用重金属如:二氧化铂、溴化钡等,降低了重金属残留污染的危险,使产品符合质量标准。
改进了工艺包括如下步骤:
(1)缩合反应:肌酐与磷酸三乙酯混合,超声波震荡条件下滴加三氯氧磷。反应温度为(20±5)℃。相比专利合成方法,反应条件温和,收率高。
(2)磷酸肌酸钠粗品制备:反应时间通过液相监控肌酐磷酰氯的剩余量,确定为10~15 h。
(3)精制:专利中采用过交换树脂柱提出的到成品的方法。我们的工艺采用树脂搅拌吸附加重结晶的方法。相比专利的精制方法,我们的工艺操作简单稳定,适合工业化生产。
3 实 验
3.1 主要仪器[8-9]
LC-9A岛津高效液相色谱仪(色谱柱:C18柱,长25 cm,内径4.6 mm,填料粒径5 μm,流速1.0 mL/min,进样量20 μL),美国Agilent有限公司;流动相:0.2%磷酸二氢钾和0.1%四丁基氢氧化铵溶液(pH值为6.3);GC惠普1890Ⅱ;ARX-300型核磁共振仪(TMS内标),德国Bruker。
3.2 2-肌酐磷酰氯合成
反应釜中投入26 kg磷酸三乙酯和2.26 kg肌酸酐,超声频率为20~40 kHz,超声功率为250~350 W,(20±5)℃滴加三氯氧磷6.12 kg,约2.5 h溶液几乎澄清。加入二氯甲烷50 kg萃取磷酸三乙酯。降温至(0±5)℃,滴加20 L甲苯,搅拌1 h后,再滴加20 L甲苯,滴加完毕,(0±5)℃冷冻搅拌析晶3 h。离心,滤饼用甲苯洗先泡洗(5.0 L/次×2),离心,所得固体装袋,称湿重约6.0~6.5 kg。
3.3 磷酸肌酸钠粗品合成
反应釜中加入60 kg纯化水,5.0 kg氢氧化钠,室温下搅拌溶解。降温至(0±5)℃,开始分批加入肌酐磷酰氯(注意防潮),控制加料速度,使温度低于10 ℃,加完后逐渐升温至(10±5)℃,保温反应10~15 h,液相检测反应进度。反应结束,静置分层,将下层水层移至反应釜中,降温至0~10 ℃,滴加浓盐酸调pH=9.0~10,加入0.5 kg大孔树脂,室温搅拌脱色1 h,加1%氢氧化钠调至pH=9.0~10,过滤,滤饼用2.0 kg纯化水洗涤。滤液转至200 L反应釜中,控制T=20~25 ℃,1 h 内滴加40.0 kg丙酮:异丙醇=2:1混合溶剂,加完后搅拌30 min,静置1 h后分液,(下层为红色油层,弃去;上层淡黄色,收集)。 向上层液中再滴加丙酮:异丙醇=2:1混合溶剂5 kg,约30 min加完,搅拌30 min,静置1 h分液。上层料液降温至0~10 ℃,搅拌2 h。开始滴加丙酮:异丙醇=2:1的混合溶剂7 kg,0~10 ℃养晶3 h,离心,滤饼用5 kg丙酮:异丙醇=2:1的混合溶剂洗涤,滤饼40~50 ℃减压干燥8 h。得磷酸肌酸钠粗品2.0~2.5 kg。
3.4 磷酸肌酸钠精制
50 L反应釜中加入20 kg纯化水,0~10 ℃搅拌溶解,用1%氢氧化钠水溶液调pH=9.0~10;加入0.1 kg大孔树脂,搅拌1 h,离心过滤。再加入活性炭0.1 kg,搅拌1 h经除碳、除菌过滤至结晶罐中。滤液中滴加约10 kg丙酮:异丙醇=2:1的混合溶剂,料液变浑,停止滴加,搅拌2 h。继续滴加10 kg丙酮:异丙醇=2:1的混合溶剂。0~10 ℃析晶2 h,离心过滤,滤饼用1 kg丙酮:异丙醇=2:1的混合溶剂洗涤,40~50 ℃ 减压干燥8 h,得产品1.2~1.5 kg。合成流程如图3所示。
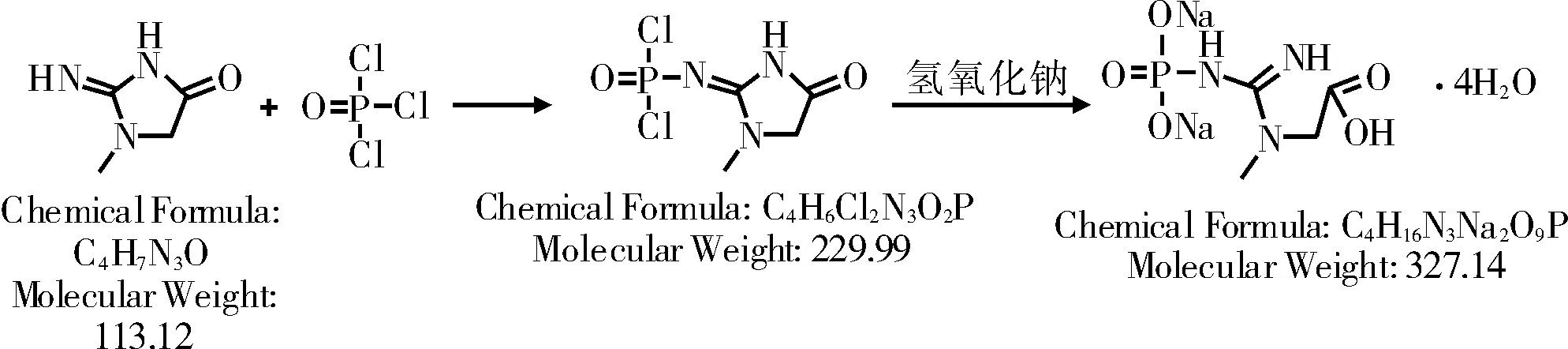
图3 磷酸肌酸钠的合成
高效液相色谱仪(色谱柱:C18柱,长25 cm,内径4.6 mm,填料粒径5 μm,Agilent);流速:1.0 mL/min;进样量:20 μL;流动相:0.2%磷酸二氢钾和0.1%四丁基氢氧化铵溶液(pH值为6.3)测试含量99.91%,水分22.3%,质量收率65%。磷酸肌酸钠结构示意图如图4所示,1H-NMR数据如表1所示。
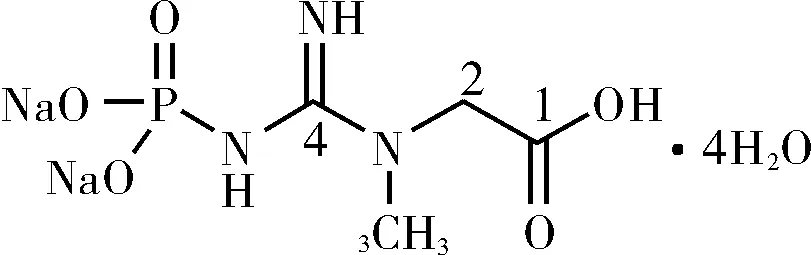
图4 磷酸肌酸钠结构
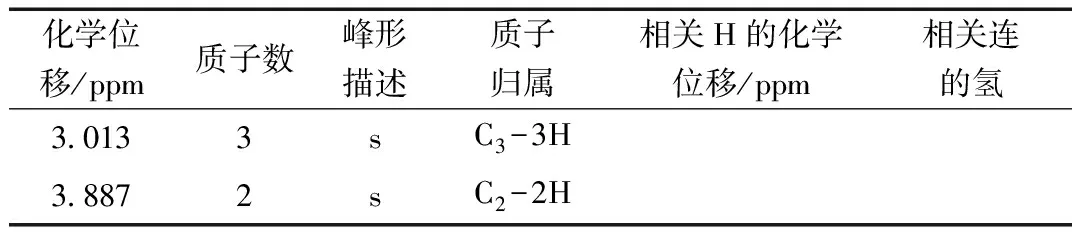
化学位移/ppm质子数峰形描述质子归属相关H的化学位移/ppm相关连的氢30133sC3-3H38872sC2-2H
供试品1H-NMR 谱显示出2组氢,共5个质子,都为单峰,其积分比(由高场到低场)约为3:2,与磷酸肌酸钠化学结构吻合。
(1)δ3.013(单峰,3H),结合本品DEPT谱和HSQC谱可知,此峰属于伯碳质子信号。在H-H COSY图谱中,未见其相关峰,应属于N-CH3的信号峰。
(2)δ3.887(单峰,2H),结合本品DEPT谱和HSQC谱可知,此峰属于仲碳质子信号。在H-H COSY图谱中,未见其相关峰,应属于N-CH2-COOH的信号峰。由于受到氨基和羧基的诱导效应,化学位移较高。
此解析结果与磷酸肌酸钠结构相符,供试品与对照品氢谱基本一致。
4 结果与讨论
在肌酐磷酰氯的制备过程中,磷酸三乙酯和肌酸酐在超声条件下混合,超声频率为20~ 40 kHz,超声功率为250~350 W,(20±5)℃滴加三氯氧磷。文献[7]条件为肌酸酐和三氯氧磷在回流条件108 ℃下反应。相对于回流反应温度,制备为常温,更容易控制,反应在超声条件下进行,反应更完全,收率也更高。
在制备过程中增加了利用高效液相监控肌酐磷酰氯的剩余量,确定反应时间为10~15 h。
从试验结果看,结晶条件确定为大孔树脂搅拌吸附加活性炭过滤重结晶,比树脂纯化工艺简单,结晶时间短,所得产品杂质低,综合产品稳定性和实际生产情况,确定了大孔树脂搅拌吸附加活性炭吸附。
对磷酸肌酸钠晶型进行研究,发现溶剂、搅拌速度、搅拌时间对其有重要影响。通过试验验证,确定了晶型溶剂为丙酮和异丙醇混合溶剂,采用比例为丙酮:异丙醇=2:1,在析晶过程中快速搅拌8 h得到药用晶型。经检验各项指标均符合2015版药典质量标准。
5 结 论
(1)最佳条件确定为:磷酸三乙酯和肌酸酐在超声条件下混合,超声频率为20~40 kHz,超声功率为250~350 W,(20±5)℃滴加三氯氧磷制备肌酐磷酰氯,在粗品的制备过程中液相监控肌酐磷酰氯的剩余量,监控时间为10~15 h,结晶条件为大孔树脂吸附然后加活性炭过滤,结晶溶剂为丙酮:异丙醇=2:1,结晶时间为8 h。
(2)测试含量99.91%,水分22.3%,质量收率65%。
(3)通过这次的工艺优化和3批生产验证,表明该工艺适合规模化生产,反应条件可控,经检验各项指标均符合2015版药典质量标准。
[1] 王景辉,李梦青,刘桂敏,等. 磷酸肌酸的应用研究[J].天津药学,2004,16(1):60-62.
[2] Karlzeile H, Gard M. Creatine phosphoric acid: a second Method of Synthesis[J]. Z.Physiol chem,1938,256:131-140.
[3] 候立向.一种磷酸肌酸钠的生产方法[P].中国:1478899.
[4] SHIVER HE. Physicoch emistry of creatine and Creatinime[J]. Chem.Reviews,1929,6: 419-444.
[5] 赵春山.磷酸肌酸钠的合成研究[J].哈尔滨理工大学报,2004,9(4):124-126.
[6] 汤磊,王建塔,朱高峰.磷酸肌酸二钠的制备[J].中国医药工业杂志,2009,40(3):172-173.
[7] Pierre MJO.Process for the preparation of 2-dichlorophosphpryl-creatinine[P].US,3632603,1972-01.
[8] 国家药典委员会.中国药典(二部)[S].北京:中国医药科技出版社,2015:172-176.
[9] 张晓平,米沙,杨建国.高效液相色谱法测定磷酸肌酸钠含量及其应用[J].天津药学,2003,15(1):18-20.
Synthesis of Phosphocreatine Disodium
CHENNing
(Hainan Zhonghua United Medicine Industry Co., Ltd., Hainan Haikou 570216, China)
After researched in the synthesis of phosphocreatine disodium, the synthesis routine of phosphocreatine disodium was confirmed and can be used for industrial production. Compared with other routes, the process conditions had the advantage of initial reactants, mild reaction conditions, high yield and simple operation. With creatinine as raw material, mix with triethyl phosphate, and then added phosphorus oxychloride under the condition of ultrasound, phosphoccreatinine disodium resulted from the condensation reaction, and then cleavaged of the lactam cycle in the presence of sodroxide. The crude products stired with macroporous resin adsorption. Phosphocreatine sodium resulted from the recrystallization. Structure was confirmed by the1H-NMR. Phosphocreatine disodium was in good yield (65%), content was more than 99.9%. The quality of phosphocreatine disodium can meet the requirement of CP2015.
phosphocreatine disodium; creatinine; synthesis
陈宁(1981-),男,本科,工程师,主要从事原料药研发工艺研究。
R972
A
1001-9677(2016)023-0048-03
