高通量基因组测序在农作物基因定位与发掘中的应用
2017-01-12余新桥张安宁王飞名刘国兰
刘 毅,余新桥,张安宁,王飞名,刘国兰
(上海市农业生物基因中心,上海 201106)
高通量基因组测序在农作物基因定位与发掘中的应用
刘 毅,余新桥,张安宁,王飞名,刘国兰*
(上海市农业生物基因中心,上海 201106)
随着新一代测序技术的发展和测序成本的不断降低,高通量测序在植物研究领域中得到广泛应用。通过简要阐述近年来高通量基因组测序在农作物研究中的应用进展,重点介绍了基于基因组重测序的作物基因定位与发掘新方法。这些将为农作物新品种选育和改良带来新思路,极大地缩短育种进程。
高通量测序;基因组;农作物;基因定位
高通量测序(High-throughput sequencing)的特点是数据量大、成本低,一次可以对上百万条DNA分子序列进行测定,也被称为第二代测序技术(Next generation sequencing),它是核酸研究的一次革命技术创新,为功能基因组学研究带来了新的科研方法和解决方案。
1 测序技术的发展
1977年,几乎是在同一时期Maxam等[1]和Sanger等[2]两组科研人员分别发表了通过化学降解测定DNA序列的方法和利用末端终止反应的DNA测序方法(Sanger测序法)。以Sanger测序法为代表的第一代测序技术极大促进了生物科学的研究,然而也存在成本高、通量低、消耗时间长等缺点,因此以大数据、低成本为特征的高通量测序技术发展迅速,其中包括Roche公司的454测序仪,Illumina公司的Solexa基因组分析仪和ABI公司的SOLiD测序仪[3-6]等测序平台,其中Illumina测序技术具有高准确性、高通量、高灵敏度和低运行成本等优势,在基因组测序研究中应用最为广泛。目前第三代测序技术已经开发应用,并逐步被认识和利用,称为单分子测序[7]。
在植物研究方面,由于高通量测序技术的出现,全基因组测序所需的时间与成本均大幅下降,使对一个物种基因组进行细致全貌的分析成为可能,从而带动了植物育种研究应用开始进入分子水平,已成为挖掘与农作物抗性、产量、品质等优异性状相关候选基因的重要手段。
2 全基因组De novo测序
De novo测序可获得任何一个物种的基因组序列图谱,进而构建该物种的基因组数据库,推动下游一系列研究工作的展开。2000年,模式植物拟南芥(Arabidopsis thaliana)利用传统的Sanger法完成全基因组测序[8],随后水稻(Oryza sativa)全基因组序列于2002年发表[9],人类基因组计划与多种重要植物基因组计划也相继顺利完成[10-16]。随着测序技术的发展,研究人员开始选择第二代测序技术进行全基因组De novo测序,熊猫(Ailuropoda melanoleura)基因组测序工作是大型物种中第一个使用新一代测序技术完成的。黄瓜(Cucumis sativus)是第一个完成全基因组测序的蔬菜作物,近年来利用高通量测序技术,已经有包括马铃薯(Solanum tuberosum)、小麦(Triticum aestivum)、油菜(Brassica napus)、棉花(Gossypium raimondii)等主要农作物基因组测序完成[17-20](http://www.ncbi.nlm.nih.gov/)。和传统技术比较,高通量测序所需的成本和时间都大大的降低,大规模物种全基因组De novo测序将会越来越多,基因组学研究也将进入一个新的时期。
3 基因组重测序
基因组重测序是指对已有参考基因组序列的物种进行个体或群体的全基因组测序,可以获得基因组上单核苷酸多态性位点(SNP,single nucleotide polymorphism)、插入缺失(InDel,insertion-deletion)和结构变异(SV,structural variation)等遗传特征,为遗传学研究和分子育种提供多态性标记信息,为该物种进化、驯化过程以及功能基因组学的研究提供海量数据。
Lai等[21]对包括Zheng58,5003,478,178,Chang7—2和Mo17在内的6个国内重要的玉米杂交组合骨干亲本进行了全基因组重测序,结果得到了100多万个SNPs和3万多个InDels,建立了高密度的分子标记遗传图谱,并在101个低序列多态性区段中发现有大量候选基因与玉米的选育改良相关。Branca等[22]对26个蒺藜苜蓿材料进行了全基因测序,分析其序列差异和连锁不平衡(LD,linkage disequilibrium)情况,检测得到约300万个SNPs,有利于在全基因组关联分析中定位豆科植物中共生关系和结瘤相关性状的位点。Zheng等[23]通过对3个高粱品种的全基因组重测序,发现在籽实高粱和甜高粱间约有1 000多个基因存在序列和结构的差异,涉及到糖与淀粉代谢、木质素和香豆素合成、核酸代谢、胁迫响应和DNA修复等过程。Lin等[24]对来自全世界的360份番茄进行了重测序,发现了1 100多万个SNPs,构建了番茄的变异图谱,为番茄的全基因组分子育种提供了基础。Lam等[25]比对野生大豆和栽培大豆重测序数据比较得到630多万个SNPs,发现了大量在栽培大豆中获得以及丢失的野生大豆基因。Qi等[26]通过重测序构建了黄瓜遗传变异图谱,发掘了2 000多个在驯化过程中受选择的基因。Zhang等[27]利用上述数据进行了黄瓜大片段DNA序列的结构变异鉴定和分析,揭示了SV产生的主要机制。Xu等[28]通过对40个代表性水稻品种和10个野生稻资源的基因组重测序,同样发现了数千个与水稻人工选择相关的基因,并且证明籼稻和粳稻的起源是相互独立的,而粳稻是从中国普通野生稻进化而来。中国农业科学院的3K水稻基因组计划已经收集了全世界不同遗传背景和表型的2 859份水稻资源,并通过重测序获得了基因组信息,平均覆盖深度为14倍,建立了水稻基因组的SNP与InDel变异数据库[29-30]。Huang等[31]利用446份普通野生稻和1 083个栽培稻重测序数据,构建了水稻基因组变异图谱并确定了55个在驯化过程中发生的选择性清除(Selective sweep),进一步分析揭示了粳稻最先起源于中国南方,籼稻是随后由粳稻与当地野生稻杂交形成的。这些结果将有效指导和加速农作物分子育种等研究,具有重要的科研价值和产业价值。
4 高通量测序技术与基因挖掘
高通量测序极大地促进了重要功能基因或者数量性状位点(QTL,quantitative trait locus)和分子标记的挖掘,在重测序获得的大量遗传变异基础上,通过关联分析、比较研究、连锁分析和生物信息学分析可以开发出高密度的分子标记,能显著促进种质资源挖掘和利用,缩短育种周期(图1)。
4.1 重测序应用于GWAS
全基因关联分析(GWAS,Genome-wide association study)最早在医学研究中被广泛。GWAS分析无需在研究前构建任何假设,可以将目标表型与其DNA序列变异关联,分析的信息量大,能检测到大量与目标性状相关的基因。基于测序来确定关键位点的GWAS分析方法,与传统的QTL定位方法互为补充。
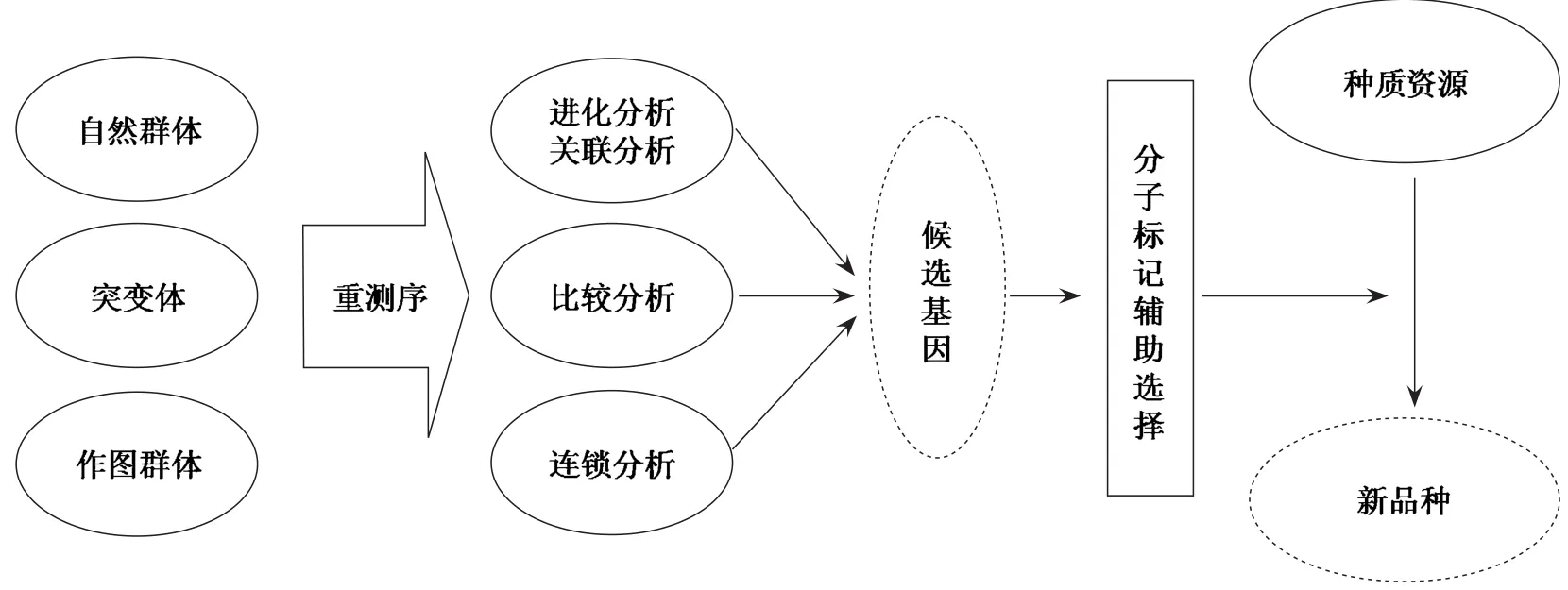
图1 基于高通量测序技术的基因发掘研究思路Fig.1 The research approach of plant gene discovery based on high-throughput sequencing

表1 高通量基因组测序在水稻研究中的应用Table 1 Application of high-throughput genome sequencing in rice research
Tian等[32]通过GWAS分析获得一个玉米嵌套的关联映射面板,利用160万个SNP位点鉴定了与茎叶夹角相关的基因。Huang[33]等通过新的基因型分类方法,构建了517个水稻地方品种的高密度的单体型图谱,然后利用360万个SNP位点对373个籼稻群体的对包括株型、产量、米质和生理生化等14个农艺性状进行GWAS研究,发现有6个位点与前人研究结果相符。随后,Huang等[34]在这些数据的基础上,增加了430份来自世界不同地区的水稻材料的重测序数据,对这些品种的抽穗期和产量性状进行了GWAS研究,定位了32个新的抽穗、产量性状相关的位点。Jia等[35]构建一张包含国内外916份谷子品种的高精度单倍体型图谱,通过GWAS分析定位到了512个与株型、产量、花期、抗病性等47个农艺性状关联的位点,同时还发现了36个在新品种选育中受选择的特殊位点。
4.2 重测序应用于突变体
突变体的表型和基因型为基因功能研究提供了直接的证据,而采用全基因组重测序来鉴定突变体的变异序列已经成为功能基因组学研究的一个发展趋势。
主要农艺性状是由多基因控制的,而单个基因仅只有很小的表型效应,因此鉴定和克隆单突变位点非常困难。Abe[36]介绍一种快速基因定位方法——Mutmap法,用突变体与野生型杂交构建F2分离群体的,在分离群体构建基因池进行全基因组重测序。Abe构建了7个分离群体,其中一个亲本是日本骨干水稻栽品种,鉴定了包括控制淡绿色叶片和半矮生性状相关突变基因位点。这种方法只需在F2分离群体中选择极端性状构建等基因池,利用高通量测序手段发掘与性状相关联的SNP位点,得到候选基因,为农作物基因克隆研究提供了新思路,减少了在传统图位克隆中构建群体所耗费的时间。
陈竹锋等[37]利用改进的MutMap方法成功鉴定了LOC-Os02 g40450(MER3)是控制osms55突变体雄性不育的基因。改进的MutMap方法不需要组装野生型基因组序列,而是分别将两个混池的重测序数据与‘日本晴’参考基因组进行比对,通过比较突变体和野生型的差异SNP确定候选基因,节约了成本。Takagi等[38]2015年对日本当地品种‘Hitomebore’进行EMS诱变处理,产生耐盐性的突变品系(hst1)。运用MutMap方法,对20个F2子代群体极端性状混池和测序,通过差异分析,成功找到水稻耐盐性突变体产生机制,由于OsRR22基因中插入了Tos17基因,导致OsRR22基因部分功能缺失,产生耐盐性。
4.3 重测序应用于遗传图谱
传统的QTL定位所用的分子标记连锁遗传图谱密度不高,定位得到的性状相关位点不够准确。新一代测序技术可以提供大量的可靠数据,使构建超高密度的遗传图谱成为可能。
Wang等[39]构建了一个包含2 334个SNP的水稻重组自交系遗传图谱,鉴定到了49个与14个农艺性状相关的QTL,其中分别控制分蘖角度、株高、剑叶宽度、粒长、粒宽的5个主效QTL的遗传距离均很小,直接可以分析其区间内的候选基因。Yu等[40]为了评估所构建的‘珍汕97’和‘明恢63’重组自交系遗传图谱的质量,验证了包括GS3、GW5和OsC1这3个分别控制粒长、粒宽和色泽的主效QTLs位点,被选目标位点都被精确地定位在基因实际所在的bins中,表明其获得的遗传图谱准确性很高。Takagi[41]提出了一种应用重测序方法进行QTL定位的新技术(QTL-seq法),利用目标性状差异较大的一对品种杂交后获得的重组自交系或F2,在群体中选择极端性状的20—50个个体分别构建高低表型DNA混池后进行重测序,通过对比两个混池的SNP位点的测序深度相关的一个参数(SNP-index)来定位QTL。文中以水稻为例对该方法用了两种分离群体进行了验证,并对此方法的运用进行了数据模拟,确定混池所选极端个体数目占群体总数的比例及测序深度是限制因素,发现当所选个体比例为15%,测序深度在20×时,此方法可应用于F2代群体进行QTL的准确定位,如果应用在F7代的RILs群体,则定位效果更显著。Yang等[42]对Nipponbare×LPBG的F3后代(10 800个体)进行苗期耐冷性的QTL定位,分别选取430个极端敏感个体和385个极端耐冷个体构建混池,对两个DNA混池测序得到45万个SNPs,通过两种分析方法均定位到6个主效QTLs,位于第1,2,5,8和10染色体上,和前人定位区间相近。Lu等[43]利用QTL-seq的技术思路,在黄瓜中快速定位了一个与早开花相关的QTL。通过QTL-seq定位和遗传图谱QTL分析结合将性状相关区段缩短至第1染色体上的25.42—26.31Mb,根据注释在该区域中,包含基因Csa1G651710,与拟南芥的FT基因同源性高达74%。Illa等[44]利用6个F2群体,采用QTL-seq方法快速定位到了3个新的番茄果实重量位点和3个控制心室数量的位点,进一步通过精细定位将果重的一个主效QTL(fw11.2)缩短至750 kb范围内,其中包含66个候选基因。
5 展望
基于高通量测序技术的基因定位与传统方法相比周期短、效率高;所获标记数量多、密度高;定位准确性强、精度高,可经济高效地实现功能基因定位,利用快速而精确的分子标记辅助选择,在早世代进行标记选择,还能对一些难以精确评价的抗逆性表型(如抗旱性等)进行筛选,将显著提高育种效率。可以预期的是,随着高通量测序技术的不断发展和完善,试验成本的逐步降低,基于高通量测序手段的基因分型和分子标记开发以及候选功能基因的挖掘,将会更加广泛地应用于农作物分子育种研究中,加速农业育种研究进程。
[1]MAXAM A M,GILBERT W.A New Method for Sequencing DNA[J].Proceedings of the National Academy of Sciences,1977,74(2):560-564.
[2]SANGER F,NICKLEN S,COULSON A R.DNA Sequencing with Chain-Terminating Inhibitors[J].Proceedings of the National Academy of Sciences,1977,74(12):5463-5467.
[3]MARGULIES M,EGHOLM M,ALTMAN W E,et al.Genome sequencing in microfabricated high-density picolitre reactors[J].Nature,2005,437(7057):376-380.
[4]SHAFFER C.Next-generation sequencing outpaces expectations[J].Nature Biotechnology,2007,25(2):149.
[5]SHENDURE J,JI H.Next-generation DNA sequencing[J].Nature Biotechnology,2008,26(10):1135-1145.
[6]SCHUSTER S C.Next-generation sequencing transforms today's biology[J].Nature Methods,2008,5(1):16-18.
[7]EID J,FEHR A,GRAY J,et al.Real-Time DNA Sequencing from Single Polymerase Molecules[J].Science,2009,323(5910):133-138.
[8]TAGI.Analysis of the genome sequence of the flowering plant Arabidopsis thaliana[J].Nature,2000,408(6814):796-815.
[9]YU J,HU S,WANG J,et al.A draft sequence of the rice genome(Oryza sativa L.ssp.indica)[J].Science,2002,296(5):79-92.
[10]TUSKAN G A,DIFAZIO S,JANSSON S,et al.The genome of black cottonwood,Populus trichocarpa(Torr.&Gray)[J].Science,2006,313(5793):1596-1604.
[11]JAILLON O,AURY J M,NOEL B,et al.The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla[J]. Nature,2007,449(7161):463-467.
[12]MING R,HOU S,FENG Y,et al.The draft genome of the transgenic tropical fruit tree papaya(Carica papaya Linnaeus)[J].Nature,2008,452(7190):991-996.
[13]ZHARKIKH A,TROGGIO M,PRUSS D,et al.Sequencing and assembly of highly heterozygous genome of Vitis vinifera L.cv Pinot Noir:Problems and solutions[J].Journal of Biotechnology,2008,136(1-2):38-43.
[14]PATERSON A H,BOWERS J E,BRUGGMANN R,et al.The Sorghum bicolor genome and the diversification of grasses[J].Nature,2009,457(7229):551-556.
[15]SCHNABLE P S,WARE D,FULTON R S,et al.The B73 maize genome:Complexity,diversity,and dynamics[J].Science,2009,326(5956):1112-1115.
[16]SCHMUTZ J,CANNON S B,SCHLUETER J,et al.Genome sequence of the palaeopolyploid soybean[J].Nature,2010,463(7278):178-183.
[17]Potato Genome Sequencing Consortium.Genome sequence and analysis of the tuber crop potato[J].Nature,2011,475(7355):189-195.
[18]HUANG S,LI R,ZHANG Z,LI L,et al.The genome of the cucumber,Cucumis sativus L[J].Nature Genetics,2009,41(12):1275-1281.
[19]LING H Q,ZHAO S,LIU D,WANG J,et al.Draft genome of the wheat A-genome progenitor Triticum urartu[J].Nature,2013,496(7443):87-90.
[20]CHALHOUB B,DENOEUD F,LIU S,et al.Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome[J].Science,2014,345(6199):950-953.
[21]LAI J,LI R,XU X,JIN W,et al.Genome-wide patterns of genetic variation among elite maize inbred lines[J].Nature Genetics,2010,42(11):1027-1030.
[22]BRANCA A,PAAPE T D,ZHOU P,et al.Whole-genome nucleotide diversity,recombination,and linkage disequilibrium in the model legume Medicago truncatula[J].Proceedings of the National Academy of Sciences,2011,108(42):E864-70.
[23]ZHENG LY,GUO X S,HE B,et al.Genome-wide patterns of genetic variation in sweet and grain sorghum(Sorghum bicolor)[J].Genome Biology,2011,12(11):R114.
[24]LIN T,ZHU G,ZHANG J,et al.Genomic analyses provide insights into the history of tomato breeding[J].Nature Genetics,2014,46(11):1220-1226.
[25]LAM H M,XU X,LIU X,et al.Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection[J].Nature Genetics,2010,42(12):1053-1059.
[26]QI J,LIU X,SHEN D,et al.A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity[J].Nature genetics,2013,45(12):1510-1515.
[27]ZHANG Z,MAO L,CHEN H,et al.Genome-Wide Mapping of Structural Variations Reveals a Copy Number Variant That Determines Reproductive Morphology in Cucumber[J].The Plant Cell,2015:tpc.114.135848.
[28]XU X,LIU X,GE S,et al.Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes[J].Nature biotechnology,2012,30(1):105-111.
[29]LI J Y,WANG J,ZEIGLER R S.The 3,000 rice genomes project:new opportunities and challenges for future rice research[J].GigaScience,2014,3(1):1-3.
[30]郑天清,余泓,张洪亮,等.水稻功能基因组育种数据库(RFGB):3K水稻SNP与InDel子数据库[J].科学通报,2015,60(4):367-371.
[31]HUANG X,KURATA N,WEI X,et al.A map of rice genome variation reveals the origin of cultivated rice[J].Nature,2012,490(7421):497-501.
[32]TIAN F,BRADBURY P J,BROWN P J,et al.Genome-wide association study of leaf architecture in the maize nested association mapping population.Nature Genetics,2011,43(2):159-162.
[33]HUANG X,WEI X,SANG T,et al.Genome-wide association studies of 14 agronomic traits in rice landraces[J].Nature Genetics,2010,42(11):961-967.
[34]HUANG X,ZHAO Y,WEI X,et al.Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm[J].Nature genetics,2012,44(1):32-39.
[35]JIA G Q,HUANG X H,ZHI H,et al.A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet(Setaria italica)[J].Nature Genetics,2013,45(8):957-961.
[36]ABE A,KOSUGI S,YOSHIDA K,et al.Genome sequencing reveals agronomically important loci in rice using MutMap[J].Nature Biotechnology,2012,30(2):174-178.
[37]陈竹锋,严维,王娜,等.利用改进的MutMap方法克隆水稻雄性不育基因[J].遗传,2014,36(1):85-93.
[38]TAKAGI H,TAMIRU M,ABE A,et al.MutMap accelerates breeding of a salt-tolerant rice cultivar[J].Nature biotechnology,2015,33(5):445-449.
[39]WANG L,WANG A,HUANG X,et al.Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines[J].Theoretical and applied genetics,2011,122(2):327-340.
[40]YU H,XIE W,WANG J,et al.Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers[J].PLoS One,2011,6(3):e17595.
[41]TAKAGI H,ABE A,YOSHIDA K,et al.QTL-seq:rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations[J].Plant Journal,2013,74(1):174-183.
[42]YANG Z,HUANG D,TANG W,et al.Mapping of Quantitative Trait Loci Underlying Cold Tolerance in Rice Seedlings via High-Throughput Sequencing of Pooled Extremes[J].PLoS One,2013,8(7):670-692.
[43]LU H,LIN T,KLEIN J,et al.QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber[J].Theoretical and Applied Genetics,2014,127(7):1491-1499.
[44]ILLA BERENGUER E,VAN HOUTEN J,HUANG Z,et al.Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq[J].Theoretical and Applied Genetics,2015,128(7):1329-1342.
(责任编辑:张睿)
Application of high-throughput genome sequencing in crop gene discovery and mapping
LIU Yi,YU Xin-qiao,ZHANG An-ning,WANG Fei-ming,LIU Guo-lan*
(Shanghai Agrobiological Gene Center,Shanghai 201106,China)
High-throughput sequencing,which has an ultra-low cost per base of sequencing and an overwhelmingly high data output,has been widely used in the field of plant research.In this paper,we review the progress in the application of high-throughput sequencing technologies to crop research,especially the new methods in crop gene discovery and mapping.These novel research methods and solutions will greatly shorten the breeding process.
High-throughput Sequencing;Genome;Crop;Gene mapping
S188.1
A
1000-3924(2016)06-171-05
2015-10-26
国家高技术研究发展计划(863计划)项目(2014AA10A604);上海市市级农口系统青年人才成长计划[沪农青字(2015)第1-6号]
刘毅(1984—),男,在职博士,助理研究员,研究方向:节水抗旱稻分子育种。E-mail:ly07@sagc.org.cn
*通信作者,E-mail:lgl@sagc.org.cn
