H2O2/[Hnmp]HCOO氧化苯并噻吩类硫化物的量子化学研究*
2017-01-10王寒露陈华锴
王寒露,周 臻,陈华锴
(广东石油化工学院,化学工程学院,广东 茂名 525000)
H2O2/[Hnmp]HCOO氧化苯并噻吩类硫化物的量子化学研究*
王寒露†,周 臻,陈华锴
(广东石油化工学院,化学工程学院,广东 茂名 525000)
针对质子型离子液体[Hnmp]HCOO在加入H2O2后生成的过氧化物[Hnmp]HCOOO氧化苯并噻吩(BT)、二苯并噻吩(DBT)和4,6-二甲基二苯并噻吩(4,6-DMDBT)的氧化脱硫反应,采用密度泛函理论DFT/B3LYP-D3方法考察了这三种硫化物的反应历程。首先通过几何优化得到了[Hnmp]HCOO和[Hnmp]HCOOO的稳定结构,然后考察了[Hnmp]HCOOO氧化BT、DBT和4,6-DMDBT的反应机理,寻找可能的过渡态、中间体,计算各步反应的能量变化。结果表明,[Hnmp]HCOOO先将硫化物氧化成亚砜,然后进一步氧化成砜;在氧化过程中,DBT的反应速率是三者中最快的,与文献的实验结果一致,且H2O2/[Hnmp]HCOO体系的脱硫效果优于传统的H2O2/HCOOH体系。
离子液体;硫化物;密度泛函理论;氧化萃取脱硫
0 前 言
SO2的排放对环境造成了巨大的污染,为了人类和经济社会的可持续发展,世界各国对成品油的含硫量做了严格的规定[1]。石油加工业中采用传统的加氢脱硫(hydrodesulfurization,HDS)工艺虽然可以脱除硫醇和硫醚等硫化物,但是难以脱除噻吩及其衍生物[2]。为了解决HDS工艺的不足,研究者们开始将目光转移到其他脱硫方法的开发[3-6]。BOSMANN等[7]首先发现离子液体能脱除燃油中的硫化物。随后LO等发现离子液体氧化萃取脱硫方法能除去油品中大部分的硫化物[8-10],比单独的萃取脱硫更为有效。为了进一步探索离子液体的反应机制,ANANTHARAJ等[11-17]用量子化学方法模拟了咪唑类离子液体与含硫化合物的相互作用。吕仁庆等[18]模拟了吡啶基离子液体与噻吩类化合物的相互作用。2014年,LÜ等[19]用N-甲基吡咯烷酮(N-methyl-2-pyrrolidone,nmp)与甲酸(HCOOH)制备了质子型离子液体 [Hnmp]HCOO,并考察了其加入H2O2后的氧化萃取脱硫作用,结果表明该体系具有高效的脱硫性能和良好的循环利用性,并提出了[Hnmp]HCOO/H2O2与DBT的作用机理。与其他离子液体作用机制不同,[Hnmp]HCOO易与H2O2反应生成过氧化物[Hnmp]HCOOO,从而进一步氧化含硫化合物。本文采用DFT/B3LYP[20-21]并结合D3对弱相互作用能进行校正,考察了[Hnmp]HCOOO与BT、DBT和4,6-DMDBT的反应历程,拟对深入开发功能化离子液体提供参考。
1 计算模型及方法
采用密度泛函理论B3LYP/6-31G(d) 优化并筛选出稳定的[Hnmp]HCOO的结构。优化了[Hnmp]HCOO与H2O2气相反应体系中的过渡态、中间体和产物[Hnmp]HCOOO的结构,同时也优化了[Hnmp]HCOOO和BT、DBT、4,6-DMDBT气相反应体系中各步反应的反应物、过渡态、中间体和产物的结构,并进行了单点能的计算和零点能的校正。计算了所有被优化的几何结构的振动频率,确保所有的驻点(反应物、中间体和产物)没有虚频,过渡态有唯一的虚频。由于标准的B3LYP泛函不能准确地描述弱相互作用,因此在6-311++g(d,p)水平上进行了色散校正的B3LYP-D3单点计算[22]。上述计算均在Gaussian09[23]程序包上进行。结合能使用公式(1)计算:

其中Ecomplex是复合物的能量,EIL是离子对的能量,Ex是与离子对结合的物质的能量。
2 结果与讨论
以氧化DBT为例。前人的研究结果指出有机酸催化剂的氧化脱硫与过氧酸的形成有关[24-26],李连峰等[27]用HCOOH与H2O2制备HCOOOH,实验结果表明该反应在室温能快速进行,据此,LÜ等[19]结合实验数据得出[Hnmp]HCOO离子液体的氧化机理。反应步骤如下:

在优化离子对结构时,分别单独优化了[Hnmp]+和[HCOO]-。在设计[Hnmp]HCOO离子液体时,将阳离子正电性较高的区域靠近阴离子负电性较高的区域。H原子不仅可以与N原子、O原子形成氢键,还与C原子有弱相互作用,虽然这种相互作用比较弱,但对分子的性质影响还是比较显著的,尤其是这种弱相互作用可被电荷辅助作用加强,因此在设定初始结构的时候也将这种弱相互作用考虑进去。通过DFT/B3LYP方法优化得到能量最低的稳定构型,也优化了BT、DBT和4,6-DMDBT,其分子构型及结构参数见图1。
前线分子轨道,即能量最高的电子占有轨道(最高占据轨道,HOMO)和能量最低的电子未占轨道(最低空轨道,LUMO)。分子进行化学反应时,反应活性与前线分子轨道有关,因此其具有特殊地位。图2列出了[Hnmp]HCOO、[Hnmp]HCOOO和硫化物的前线分子轨道。硫化物的HOMO都是成键轨道,LUMO是反键轨道。[Hnmp]HCOO的HOMO布局在[Hnmp]上,而LUMO布局在HCOO上。[Hnmp]HCOOO的HOMO和LUMO都布局在HCOOO上。LUMO和HOMO能量级差(能隙)能反映出分子的反应活性。能隙越小,分子越不稳定,反应活性越大。[Hnmp]HCOOO的能级差(4.2 eV)要远小于[Hnmp]HCOO(7.0 eV),因此[Hnmp]HCOOO的反应活性更强。三种硫化物中,BT的能隙最大,说明其反应活性较DBT和4,6-DMDBT要低。

图1 分子结构及键长(Å)Fig.1 Molecular structures and bond lengths ( Å)

图2 [Hnmp]HCOO、[Hnmp]HCOOO和硫化物的前线分子轨道图Fig.2 The frontier molecular orbitals of [Hnmp]HCOO,[Hnmp]HCOOO and the sulfides
图3是BT氧化反应体系中的反应物、过渡态、中间体和产物的几何结构和部分参数。反应的第一步是[Hnmp]HCOOO在范德华力的作用下靠近BT,形成复合物RC,在RC中,O2—O3之间的键长为1.446 Å,O3—S之间的键长为3.689 Å。得到的过渡态TS1有唯一虚频 295.51i cm-1,在过渡态TS1的结构中,O2—O3的键长伸长为1.624 Å,O3—S之间的键长缩短为2.394 Å。根据频率的计算得知O3在O2和S之间来回摆动,亲核的氧原子进攻苯并噻吩上缺电子的硫原子,最终O2—O3之间的单键断裂,S与O3形成双键。从中间体IM1中可以看到S—O3的键长为1.520 Å,即已形成苯并噻吩亚砜(BTO)。然而生成的BTO并不是最稳定的物质,还能继续被氧化。反应的第二步是在范德华力的作用下BTO与[Hnmp]HCOOO相互靠近形成复合物IM2从而引发反应。IM2中O3—S的距离为3.329 Å,得到的过渡态TS2有唯一的虚频427.06i cm-1,在过渡态TS2的结构中O3—S的距离则缩短为2.298 Å。从复合物IM2到过渡态TS2的过程与复合物RC到过渡态TS1经历的过程相似。[Hnmp]HCOOO的O3在S与O2之间来回摆动,亲核的氧原子进攻BTO上的缺电子的S原子,最终O2—O3之间的单键断裂,S—O3之间形成双键,放出能量形成苯并噻吩砜PC。从PC中可以看到S—O3的键长为1.466 Å,即形成苯并噻吩砜(BTO2)。

图3 BT氧化反应过程各化合物的结构及键长(Å)Fig.3 The geometries and bond lengths of the compounds in BT oxidation (unit:Å)
图4和图5是DBT和4,6-DMDBT氧化反应体系中的反应物、过渡态、中间体和产物的几何结构和部分参数。[Hnmp]HCOOO在氧化DBT的反应中得到的过渡态TS1 和TS2有唯一的虚频,分别为352.08i cm-1和437.60i cm-1。[Hnmp]HCOOO氧化4,6-DMDBT的反应中得到的过渡态TS1 和TS2有唯一虚频,分别为289.51i cm-1和387.75i cm-1。[Hnmp]HCOOO氧化DBT和4,6-DMDBT的反应历程与氧化BT的反应历程类似。从图4和图5中还可以看到TS1和TS2中S—O3的键长均为4,6-DMDBT>DBT。这是因为4,6-DMDBT中存在甲基,空间位阻增大,使得S—O3键变长。

图4 DBT氧化反应过程各化合物的结构和键长(Å)Fig.4 The geometries and bond lengths of the compounds in DBT oxidation (unit:Å)
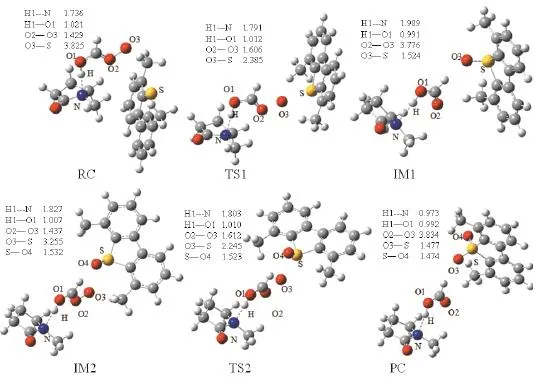
图5 4,6-DMDBT氧化反应过程各化合物的结构和键长(Å)Fig.5 The geometries and bond lengths of the compounds in 4,6-DMDBT oxidation (unit:Å)
根据计算得到的各步反应的相对电子能量得到反应的能垒图。在图6中可以清楚的看到第一步反应的能垒:BT>4,6-DMDBT>DBT,第二步反应的能垒:4,6-DMDBT>DBT>BT。BT和4,6-DMDBT的氧化反应的决速步是第一步,反应有利于生成砜类物质;而DBT的氧化反应决速步是第二步,反应有利于生成亚砜类物质。决速步能垒为BT>4,6-DMDBT>DBT,因此总的反应活性顺序为DBT>4,6-DMDBT>BT,与LÜ等[19]得出的实验结果一致。这是由于反应速率是由参与反应的原子的电子密度和空间位阻共同决定的。ZENG等[28]比较了DBT和4,6-DMDBT的S和O2、O3的净电荷。从电子密度的角度,DBT和4,6-DMDBT的硫原子的净电荷分别为0.396和0.338,给电子甲基的存在使得4,6-DMDBT上S的电子密度增加,增大了4,6-DMDBT的反应活性。
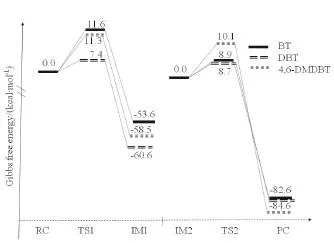
图6 BT、DBT和4,6-DMDBT氧化反应的反应势能图Fig.6 Free energy profiles of the oxidative reactions of BT,DBT and 4,6-DMDBT
3 脱硫性能评价
对比其他研究者的研究成果[29-30],发现[Hnmp]HCOO离子液体在加入H2O2后,实验中具有优良的脱硫性,主要是由于以下两个显著原因:第一,过氧酸具有很强的氧化性。ZENG等[28]计算了过氧甲酸催化氧化DBT的能垒,从DBT转变为DBTO的能垒为12.4 kcal·mol-1,从DBTO转变为DBTO2的能垒为10.1 kcal·mol-1,因此用过氧酸氧化时比用H2O2和甲酸能垒更低。可以看到在[Hnmp]HCOO离子液体中加入H2O2后反应的第一步是生成了过氧酸,因此提高了反应速率。第二,由于离子液体中存在O—H…N氢键,这些作用力使反应体系中反应物、过渡态、生成物的能垒降低,尤其是过渡态。XU等[31]计算了H2O2氧化DBT的能垒,结果显示其从DBT转变为DBTO,再从DBTO转变为DBTO2的能垒分别为32.1 kcal·mol-1和37.0 kcal·mol-1;同时计算了加入[HMIm]BF4离子液体后氧化DBT反应的能垒,发现比直接采用H2O2的能垒有大幅度的降低,两步反应的能垒分别降为13.1 kcal·mol-1和23.8 kcal·mol-1。
4 结 论
采用B3LYP-D3方法对[Hnmp]HCOO离子和H2O2参与的噻吩类化合物氧化反应的反应机理进行详细的研究。三种噻吩类芳香硫化物在离子液体中催化氧化的机理相似,但决速步不同。反应经历三个过程:[Hnmp]HCOO先生成过氧化物[Hnmp]HCOOO,然后过氧化物与硫化物反应生成亚砜,再继续被氧化生成砜。BT和4,6-DMDBT的氧化反应决速步是第一步,而DBT的氧化反应的决速步是第二步;总的反应活性顺序为:DBT>4,6-DMDBT>BT,与文献[19]的实验结果一致。分子间氢键在稳定物质结构、降低能垒方面也起到一定的作用。
[1] D’ALESSANDRO N,TONUCCI L,BONETTI M,et al.Oxidation of dibenzothiophene by hydrogen peroxide or monopersulfate and metal-sulfophthalocyanine catalysts:an easy access to biphenylsultone or 2-(2'-hydroxybiphenyl) sulfonate under mild conditions[J].New journal of chemistry,2003,27(6):989-993.DOI:10.1039/B212152B.
[2] CHU X M,HU Y F,LI J G,et al.Desulfurization of diesel fuel by extraction with [BF4]-based ionic liquids[J].Chinese journal of chemical engineering,2008,16(6):881-884.DOI:10.1016/S1004-9541(09)60010-0.
[3] 余思钰,彭晶,王寒露,等.Ti-MCM-41/次氯酸钠催化氧化脱除模型油中苯并噻吩的研究[J].新能源进展,2016,4(1):62-67.DOI:10.3969/j.issn.2095-560X.2016.01.010.
[4] BABICH I V,MOULIJN J A.Science and technology of novel processes for deep desulfurization of oil refinery streams:a review[J].Fuel,2003,82(6):607-631.DOI:10.1016/S0016-2361(02)00324-1.
[5] SONG C S,MA X L.New design approaches to ultra-clean diesel fuels by deep desulfurization and deep dearomatization[J].Applied catalysis B:environmental,2003,41(1/2):207-238.DOI:10.1016/S0926-3373(02) 00212-6.
[6] SHAFI R,HUTCHINGS G J.Hydrodesulfurization of hindered dibenzothiophenes:an overview[J].Catalysis today,2000,59(3/4):423-442.DOI:10.1016/S0920-5861(00)00308-4.
[7] BÖSMANN A,DATSEVICH L,JESS A,et al.Deep desulfurization of diesel fuel by extraction with ionic liquids[J].Chemical communication,2001(23):2494-2495.DOI:10.1039/B108411A.
[8] LO W H,YANG H Y,WEI G T.One-pot desulfurization of light oils by chemical oxidation and solvent extraction with room temperature ionic liquids[J].Green chemistry,2003,5(5):639-642.DOI:10.1039/b305993f.
[9] WANG J L,ZHAO D S,ZHOU E P,et al.Desulfurization of gasoline by extraction with N-alkylpyridinium-based ionic liquids[J].Journal of fuel chemistry and technology,2007,35(3):293-296.DOI:10.1016/ S1872-5813(07)60022-X.
[10] ZHANG C,WANG F,PAN X Y,et al.Study of extraction-oxidation desulfurization of model oil by acidic ionic liquid[J].Journal of fuel chemistry and technology,2011,39(9):689-693.DOI:10.1016/ S1872-5813(11)60041-8.
[11] ANANTHARAJ R,BANERJEE T.Phase behaviour of 1-ethyl-3-methylimidazolium thiocyanate ionic liquid with catalytic deactivated compounds and water at several temperatures:experiments and theoretical predictions[J].International journal of chemical engineering,2011,2011:209435.DOI:10.1155/2011/209435.
[12] KUMAR A A P,BANERJEE T.Thiophene separation with ionic liquids for desulphurization:a quantum chemical approach[J].Fluid phase equilibrium,2009,278(1/2):1-8.DOI:10.1016/j.fluid.2008.11.019.
[13] ANANTHARAJ R,BANERJEE T.Liquid-liquid equilibria for quaternary systems of imidazolium based ionic liquid + thiophene + pyridine + iso-octane at 298.15 K:experiments and quantum chemical predictions[J].Fluid phase equilibrium,2011,312:20-30.DOI:10.1016/j.fluid.2011.09.006.
[14] SANTIAGO R S,SANTOS G R,AZNAR M.UNIQUAC correlation of liquid-liquid equilibrium in systems involving ionic liquids:the DFT-PCM approach[J].Fluid phase equilibrium,2009,278(1/2):54-61.DOI:10.1016/ j.fluid.2009.01.002.
[15] HANKE C G,JOHANSSON A,HARPER J B,et al.Why are aromatic compounds more soluble than aliphatic compounds in dimethylimidazolium ionic liquids? A simulation study[J].Chemical physics letter,2003,374(1/2):85-90.DOI:10.1016/S0009-2614(03) 00703-6.
[16] KĘDRA-KRÓLIK K,FABRICE M,JAUBERT J N.Extraction of thiophene or pyridine from n-heptane using ionic liquids,gasoline and diesel desulfurization[J].Industrial &engineering chemistry research,2011,50(4):2296-2306.DOI:10.1021/ie101834m.
[17] ANANTHARAJ R,BANERJEE T.Quantum chemical studies on the simultaneous interaction of thiophene and pyridine with ionic liquid[J].AIChE journal,2011,57(3):749-764.DOI:10.1002/aic.12281.
[18] 吕仁庆,林进,王淑涛.吡啶基离子液体与噻吩类化合物相互作用的理论研究[J].石油学报(石油加工),2013 29(6):1015-1022.DOI:10.3969/j.issn.1001-8719.2013.06.013.
[19] LÜ H Y,WANG S N,DENG C L,et al.Oxidative desulfurization of model diesel via dual activation by a protic ionic liquid[J].Journal of hazardous materials,2014,279:220-225.DOI:10.1016/j.jhazmat.2014.07.005.
[20] SU M D,CHU S Y.Density functional study of some germylene insertion reactions[J].Journal of the American chemical society,1999,121(17):4229-4237.DOI:10.1021/ja983763j.
[21] LEE C,YANG W T,PARR R G.Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].Physical review B,1988,37(2):785-789.DOI:10.1021/ja983763j.
[22] PRAMPOLINI G,LIVOTTO P R,CACELLI I.Accuracy of quantum mechanically derived force-fields parameterized from dispersion-corrected dft data:the benzene dimer as a prototype for aromatic interactions[J].Journal of chemical theory and computation,2015,11:5182-5196.DOI:10.1021/acs.jctc.5b00642.
[23] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 09,Revision D.01[Z].Wallingford,CT:Gaussian Inc.,2013.
[24] ZICMANIS A,PAVLOVICA S,GZIBOVSKA E,et al.2-Hydroxyethylammonium carboxylates-highly biodegradable and slightly toxic ionic liquids[J].Latvian journal of chemistry,2010,49(1/4):269-277.DOI:10.2478/v10161-010-0008-6.
[25] BICAK N.A new ionic liquid:2-hydroxy ethylammonium formate[J].Journal of molecular liquids 2005,116(1):15-18.DOI:10.1016/j.molliq.2004.03.006.
[26] ANOUTI M,CAILLON-CARAVANIER M,DRIDI Y,et al.Synthesis and characterization of new pyrrolidinium based protic ionic liquids.Good and superionic liquids[J].Journal of physical chemistry B,2008,112(42):13335-13343.DOI:10.1021/jp805992b.
[27] 李连峰,樊永明,过甲酸制备过程中活性氧的转化及稳定性研究[J].中国纸业,2007,28(4):55-58.DOI:10.3969/j.issn.1007-9211.2007.04.016.
[28] ZENG X Y,MO G D,WANG H L,et al.Oxidation mechanism of dibenzothiophene compounds:A computational study[J].Computational and theoretical chemistry,2014,1037:22-27.DOI:10.1016/j.comptc.2014.03.023.
[29] ZHOU M D,MENG W Y,LI Y,et al.Extractive and catalytic oxidative desulfurization of gasoline by methyltrioxorhenium in ionic liquids[J].Energy fuels,2014,28(1):516-521.DOI:10.1021/ef402103e.
[30] LI F T,LIU R H,WEN J H,et al.Desulfurization of dibenzothiophene by chemical oxidation and solvent extraction with Me3NCH2C6H5Cl·2ZnCl2ionic liquid[J].Green chemistry,2009,11(6):883-888.DOI:10.1039/ b815575e.
[31] XU H,HAN Z,ZHANG D J,et al.Theoretical elucidation of the dual role of [HMIm]BF4ionic liquid as catalyst and extractant in the oxidative desulfurization of dibenzothiophene[J].Journal of molecular catalysis A:chemistry,2015,398:297-303.DOI:10.1016/j.molcata.2014.12.018.
Quantum Chemical Study on H2O2/[Hnmp]HCOO Ionic Liquid in the Oxidative Desulfurization of Sulfides
WANG Han-lu,ZHOU Zhen,CHEN Hua-kai
(Guangdong University of Petrochemical Technology,College of Chemistry Engineering,Guangdong Maoming 525000,China)
In this work,the oxidative desulfurization (ODS) mechanisms of benzothiophene (BT),dibenzothiophene (DBT) and 4,6-dimethyl dibenzothiophene (4,6-DMDBT) in [Hnmp]HCOO ionic liquid with H2O2as oxidant were studied by density functional theory DFT/B3LYP-D3 method.Firstly,the most stable structures of [Hnmp]HCOO and [Hnmp]HCOOO were obtained by geometrical optimization.Then the reaction mechanisms between [Hnmp]HCOOO and the three thiophene compounds were explored.The most likely transition states and intermediates were then investigated,and the energy change for each reaction step was calculated.According to the reaction mechanism,the oxidation of the thiophene compounds using peroxide [Hnmp]HCOOO had undergone processes of sulfoxide formation and sulfone formation.The results show that the ODS reaction rate of DBT is the most rapid,which is in accordance with the experimental results in reference;and the desulfurization efficiency of the H2O2/[Hnmp]HCOO system is higher than that of the traditional H2O2/HCOOH system.
ionic liquid;sulfides;DFT;oxidation extraction desulfurization
TK421;X5
A
10.3969/j.issn.2095-560X.2016.06.010
2095-560X(2016)06-0492-07
王寒露(1981-),女,博士,副教授,主要从事催化化学与计算化学研究。
2016-08-06
2016-11-23
国家自然科学基金青年基金(21403038); 广东省自然科学基金(2015A030313892);广东省高等学校优秀青年教师培养计划(YQ2015116);广东石油化工学院大学生创新创业校级培育项目(2016pyA009)
† 通信作者:王寒露,E-mail:wanghlu@mail2.sysu.edu.cn
周 臻(1994-),男,主要从事催化化学与计算化学研究。
