长效注射剂的含量和释放度测定方法的建立
2017-01-05荆广会纪新超张元元王艳娇
荆广会,杨 杰,纪新超,钟 月,张元元,王艳娇*
(沈阳药科大学 药学院,辽宁 沈阳 110016)
长效注射剂的含量和释放度测定方法的建立
荆广会,杨 杰,纪新超,钟 月,张元元,王艳娇*
(沈阳药科大学 药学院,辽宁 沈阳 110016)
目的建立用紫外可见分光光度计测定帕潘立酮棕榈酸酯长效纳米混悬注射剂的含量和体外释放度的方法。方法混悬液采用湿法研磨技术制备,含量测定溶液为四氢呋喃-N,N-二甲基甲酰胺(体积比5∶95),释放介质为质量分数0.489%聚山梨醇酯20的1 mmol·L-1盐酸溶液900 mL,检测波长为280 nm。结果在含量测定溶液中,帕潘立酮棕榈酸酯的质量浓度在20.0~60.0 mg·L-1内与吸光度呈良好的线性关系,回归方程为A=1.36×10-2ρ + 2.68×10-2(r=0.999 9),回收率在98%~102%内,精密度的RSD均小于2%(n=6);在释放介质中,帕潘立酮棕榈酸酯的质量浓度在6.0~40.0 mg·L-1内与吸光度呈良好的线性关系,回归方程为A = 1.45×10-2ρ + 9.1×10-3(r=0.999 9),帕潘立酮棕榈酸酯在释放介质中8 h内稳定。结论该方法操作简单快捷,准确性和重复性较好,可用于测定帕潘立酮棕榈酸酯制剂的含量和释放度。
药剂学;长效混悬注射剂;紫外分光光度法;帕潘立酮棕榈酸酯;含量测定;体外释放度
精神分裂症的致残率很高,在精神性障碍中十分常见,在全世界范围内的患病率达到1%左右,是最为严重的精神疾病类型。已有的流行病学研究表明,我国的精神分裂症终生的患病率有0.645%[1]。利培酮长效注射剂是第一个第二代抗精神病药的长效注射剂,帕潘立酮(9-羟基利培酮)是利培酮的活性代谢物,最初被开发为长效口服制剂[2]。帕潘立酮长效注射剂以其棕榈酸盐的形式被开发为水性纳米混悬注射液,给药方式为肌肉注射(三角肌或臀肌),给药周期为每月一针。长周期的给药方式不仅可让药物成分在患者体内持续稳定释放,保证稳定的血浆药物浓度,还能降低不良反应,使药效持续数周。此外,由于此种疗法不必每日服药,隐私保护大大增强,患者依从性大大提高,因此还可保证持续治疗,从而预防复发[3-5]。目前,帕潘立酮棕榈酸酯长效注射剂为进口制剂,其含量的测定方法主要是进口注册标准(标准号:JX20100263)中规定的HPLC法,除对色谱柱的要求高外,还需使用梯度洗脱的方式,并且流动相中含乙酸铵盐,测定起来非常麻烦。本研究旨在建立帕潘立酮棕榈酸酯含量测定的紫外分光光度法,以期为测定帕潘立酮棕榈酸酯纳米混悬液的含量和释放度提供一种快捷、简便、准确性高并且经济实用的方法。帕潘立酮棕榈酸酯(paliperidone palmitate,PP)的化学结构式见图1。
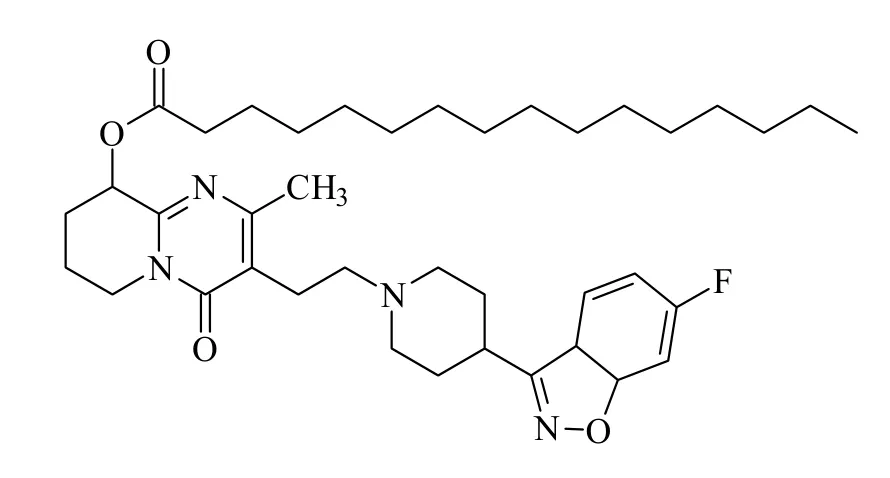
Fig. 1 The chemical structure of paliperidone palmitate图1 帕潘立酮棕榈酸酯的化学结构式
1 仪器与材料
FA1104电子分析天平(上海民桥精密科学仪器有限公司),DF-101S集热式恒温加热磁力搅拌器(巩义予华仪器制备有限公司),T6新世纪紫外可见分光光度计(北京普析通用仪器有限责任公司),耐驰实验室砂磨机 MiniZeta 03 E、0.4~0.6 mm氧化锆 ZrO2 研磨珠(NETZSCH (Shanghai) Machinery and Instruments Co., Ltd., Selb, Germany),ZRS-8G智能溶出试验仪(天津天大天发科技有限公司),BT-9300S激光粒度仪(丹东百特仪器有限公司)。
帕潘立酮棕榈酸酯(自制,含量质量分数>99%),聚山梨醇酯20(国药集团化学试剂有限公司),聚乙二醇 4000(天津柯缪化学试剂有限公司),柠檬酸(天津博迪化工股份有限公司)无水磷酸氢二钠、氢氧化钠(天津瑞金特化学品有限公司),磷酸二氢钠(西陇化工股份有限公司),娃哈哈纯净水(杭州娃哈哈集团有限公司)。
2 方法与结果
2.1 纳米混悬液的制备
称取聚山梨醇酯2.4 g、聚乙二醇4000 6.0 g、柠檬酸1 g和磷酸缓冲盐适量,加至200 mL娃哈哈纯净水中,磁力搅拌下溶解作为研磨介质。称取帕潘立酮棕榈酸酯 31.2 g,在搅拌条件下,加至研磨介质中,继续搅拌 30 min,使溶液充分浸润药物。将药物与介质溶液的初混合液加到MiniZeta研磨机中,调节转速3 000 r·min-1,控制研磨时间,使药物体积径的粒径在d(0.1)为0.3~0.6 μm,d (0.5)为0.9~1.4 μm,d(0.9)为2.0~4.4 μm内。粒径分布图见图2。
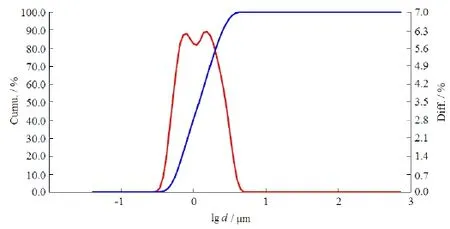
Fig. 2 The particle size distribution of paliperidone palmitate suspension injection图2 帕潘立酮棕榈酸酯纳米混悬注射液的粒径分布
光学显微镜下观察粒子在研磨前后的形态发现:研磨前帕潘立酮棕榈酸酯的原料药是层片状晶体,形状类似菱形;研磨后药物粒子大小均匀,没有聚集现象,呈类球状(图3)。一月后观察研磨液的形态和粒子状态发现,粒子略有沉降,经摇匀后测定粒径,其粒径变化不大,释放度测定结果也没有明显变化,说明所制得的研磨制剂稳定可靠(表1、2)。

Fig. 3 The shapes of paliperidone palmitate particles before(A) and after(B) grinding图3 帕潘立酮棕榈酸酯在研磨前(A)、后(B)的粒子形状变化
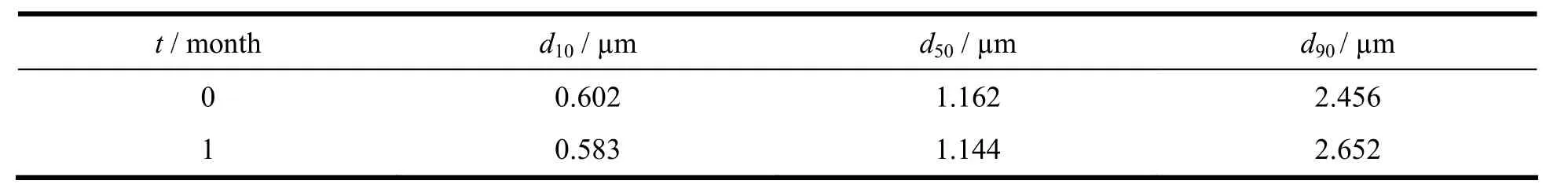
Table 1 The size of ground paliperidone palmitate particles before and after 1 month storage表1 一月前后研磨液的粒径变化

Table 2 The dissolution rate of paliperidone palmitate suspension before and after 1 month storage表2 一月前后研磨液的释放度测定结果
2.2 含量测定方法的建立
2.2.1 紫外分光光度法检测波长的确定
称取帕潘立酮棕榈酸酯20 mg,用四氢呋喃-N,N-二甲基甲酰胺(体积比5∶95)制成每1 mL约含帕潘立酮棕榈酸酯20 μg的溶液,同时配制相应比例的的辅料溶液,以空白溶剂为对照,按《中华人民共和国药典》2010年版二部附录ⅣA中紫外分光光度法测定,扫描图见图4。由图4可知,帕潘立酮棕榈酸酯在波长280 nm处有最大吸收,所用辅料在四氢呋喃-N,N-二甲基甲酰胺(体积比5∶95)溶液在280 nm处基本无干扰,故选择于波长280 nm处进行含量测定。
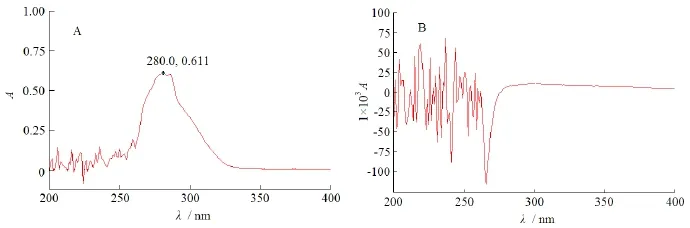
Fig. 4 The UV scanning spectra of paliperidone palmitate(A) and excipients(B) in diluent图4 帕潘立酮棕榈酸酯(A)和辅料(B)在稀释液中的紫外扫描图
2.2.2 线性关系及线性范围的确定
精密称取帕潘立酮棕榈酸酯原料药20 mg,置于50 mL量瓶中,用四氢呋喃2.5 mL振摇溶解,并用N,N-二甲基甲酰胺稀释至刻度,摇匀。再用稀释液四氢呋喃-N, N-二甲基甲酰胺(体积比5∶95)溶液分别稀释成20、30、40、50 和60 mg·L-1的帕潘立酮棕榈酸酯稀溶液,以稀释液为空白溶剂,于波长280 nm处测定吸光度值。以吸光度值(A)对质量浓度(ρ)进行线性回归,得标准曲线方程为:A=1.36×10-2ρ + 2.68×10-2(r=0.999 9)。可见帕潘立酮棕榈酸酯质量浓度在20.0~60.0 mg·L-1内与吸光度值线性关系良好。
2.2.3 稳定性试验
取质量浓度为40.0 mg·L-1的样品溶液,于室温下分别在0、2、4、6和8 h测定其吸光度,计算相对标准偏差(RSD)为0.55%。结果表明,帕潘立酮棕榈酸酯在稀释液四氢呋喃-N,N-二甲基甲酰胺(体积比5∶95)溶液中室温下8 h内稳定。
2.2.4 回收率和精密度试验
分别精密称取硝苯地平对照品32、40和48 mg各3份,置于100 mL量瓶中,分别加入相应比例的辅料,用5 mL四氢呋喃溶解并用N,N-二甲基甲酰胺定容至刻度,摇匀,滤过。取滤液1 mL置于10 mL量瓶中,用稀释液稀释至刻度,摇匀。按“2.2.1”条紫外分光光度法于波长280 nm处测定,将测得吸光度值带入“2.2.2”条下的线性方程,计算加入值,将测得值与实际加入值比较计算回收率,结果见表3。低、中、高3种质量浓度的回收率均在98%~102%内,回收率符合方法学要求。

Table 3 Recoveries of the UV method to determine PP in the diluent表3 帕潘立酮棕榈酸酯在稀释液中的紫外回收率实验结果
分别取低(30.0 mg·L-1)、中(40.0 mg·L-1)、高(50.0 mg·L-1)3种质量浓度的样品溶液,一天内重复测定5次,计算日内相对标准偏差;每天测定1次,连续测定5 d,计算日间相对标准偏差,结果见表4。
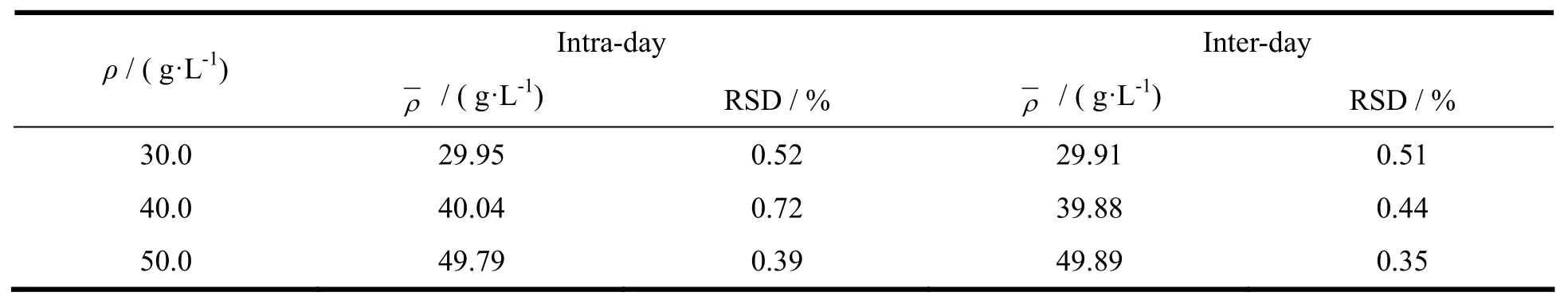
Table 4 Precision of UV method to determine PP in the diluent (n=6)表4 帕潘立酮棕榈酸酯在稀释液中的紫外精密度实验结果(n=6)
2.2.5 帕潘立酮棕榈酸酯长效注射剂的含量测定
取所得制剂样品,精密称取适量(约相当于帕潘立酮棕榈酸酯40 mg),置于100 mL量瓶中,加四氢呋喃5 mL振摇溶解,用N,N-二甲基甲酰胺稀释至刻度,摇匀。精密量取上述溶液1 mL,置于10 mL量瓶中,加稀释液稀释至刻度,摇匀,制成每1 mL稀释液中约含帕潘立酮棕榈酸酯40 µg的溶液;另取帕潘立酮棕榈酸酯原料药,同法配置制成每1 mL中约含帕潘立酮40 µg的溶液。取上述2种溶液,按《中华人民共和国药典》2010年版二部附录ⅣA紫外分光光度法,在波长280 nm处分别测定吸光度,按外标法计算,得帕潘立酮棕榈酸酯长效注射剂的含药质量分数为15.6%。
2.3 释放度测定方法的建立
2.3.1 检测波长的确定
称取帕潘立酮棕榈酸酯20 mg,置于50 mL量瓶中,加四氢呋喃2.5 mL振摇溶解,用N,N-二甲基甲酰胺稀释至刻度,摇匀。精密量取上述溶液1 mL,置于10 mL量瓶中,用释放介质(质量分数0.489%的聚山梨醇酯20的1 mmol·L-1盐酸溶液)稀释至刻度,摇匀,制成每1 mL中约含帕潘立酮棕榈酸酯40 µg的溶液。同时配制相应比例的辅料溶液,以释放介质为空白对照,按《中华人民共和国药典》2010年版二部附录ⅣA紫外分光光度法测定,扫描图见图5。由图5可知,帕潘立酮棕榈酸酯的释放介质溶液在波长280 nm处有最大吸收,所用辅料的释放介质溶液在波长280 nm处基本无干扰,故选择280 nm用于释放度的测定。
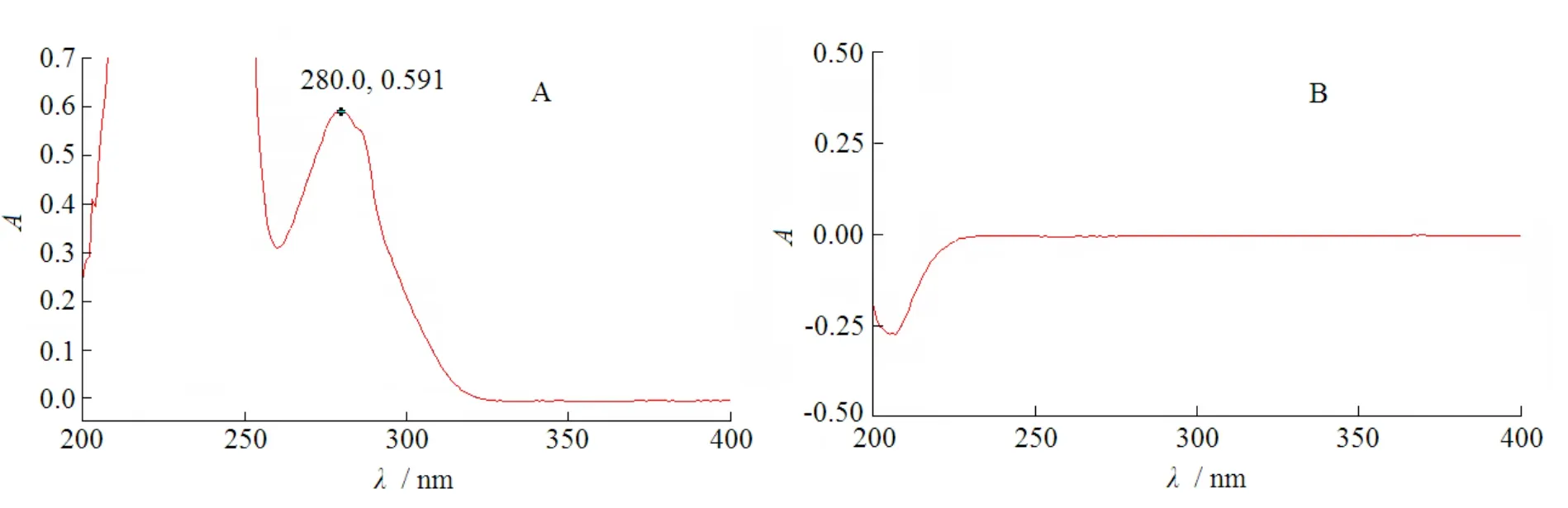
Fig. 5 The UV scanning spectra of paliperidone palmitate(A) and excipients(B) in dissolution medium图5 帕潘立酮棕榈酸酯(A)和辅料(B)在释放介质中的紫外扫描图
2.3.2 线性关系及线性范围的确定
精密称取帕潘立酮棕榈酸酯原料药20 mg,置于50 mL量瓶中,加四氢呋喃2.5 mL振摇溶解并用N,N-二甲基甲酰胺稀释至刻度,摇匀。再用释放介质分别稀释成6、10、20、30和40 mg·L-1的帕潘立酮棕榈酸酯稀溶液,以稀释液为空白,于波长280 nm处测定吸光度值。以吸光度值(A)对质量浓度(ρ)进行线性回归,得标准曲线方程为:A=1.45×10-2ρ+ 9.1×10-3(r=0.999 9)。由此可见,在释放介质中帕潘立酮棕榈酸酯的质量浓度在6.0~40.0 mg·L-1内与吸光度值线性关系良好。
2.3.3 稳定性试验
取帕潘立酮棕榈酸酯质量浓度为40.0 mg·L-1的释放介质溶液,于室温下分别在0、2、4、6和8 h测定其吸光度,计算相对标准偏差(RSD)为 0.89%。结果表明,帕潘立酮棕榈酸酯在释放介质中室温下8 h内稳定。
2.3.4 释放度的测定
帕潘立酮棕榈酸酯为进口药物,参考国家食品药品监督管理局进口药品注册标准(标准号:JX20100263),帕潘立酮棕榈酸酯长效注射液释放度测定方法确立如下:取帕潘立酮棕榈酸酯混悬液,按《美国药典)32版<711> 第二法释放度测定法,以质量分数0.489%聚山梨醇酯20的1 mmol·L-1盐酸溶液900 mL为释放介质,介质温度为(25±0.5)℃,转速为50 r·min-1,依法操作,向每个溶出杯中加入相当于(500 ± 25) µL的均匀混悬液样品。加样方式为取本品足够支数,摇匀,预先混合,将预先混合注射液约(500 ± 25) µL,置于规格为1 mL的带针头注射器内,精密称定质量,当桨转动时,将上述混悬液加入每个溶出杯中,精密称定带有针头的注射完的空注射器质量。经1.5、20和45 min时,分别取释放介质6.0 mL,立即用0.22 μm孔径的滤膜滤过,弃去初滤液至少3 mL,取续滤液3.0 mL作为供试溶液;另取棕榈酸帕利哌酮原料药40.0 mg,精密称定,置于100 mL量瓶中,加四氢呋喃5 mL振摇溶解,用N,N-二甲基甲酰胺稀释至刻度,精密量取1 mL,置于10 mL量瓶中,用释放介质稀释至刻度,摇匀,作为对照溶液。取上述2种溶液,按《中华人民共和国药典》2010年版二部附录ⅣA中紫分光光度法,在波长280 nm处分别测定吸光度,按外标一点法计算释放度。其中,由于45 min时的取样点吸光度值过高,需要用释放介质对溶液进行等量稀释后再测定。试验过程中不需要在操作容器中补充释放介质,计算公式中不需要对释放介质的体积进行校正。帕潘立酮棕榈酸酯混悬液的释放曲线见图6。
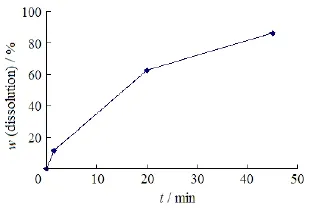
Fig. 6 The dissolution profile of paliperidone palmitate suspension injection图6 帕潘立酮棕榈酸酯混悬液的释放曲线
3 讨论与结论
a. 利用紫外分光光度法对帕潘立酮棕榈酸酯混悬注射剂进行含量和释放度测定的检测手段与进口注册标准中规定的高效液相色谱法相比,操作简便快捷,节约有机试剂,同时准确度和精密度等符合要求。
b. 对混悬液进行含量和释放度检测时,要取5支以上注射剂进行混合,充分摇匀后取样以避免因为药物分布不均造成结果的偏离。
[1] 秦群生. 精神分裂症的药物治疗现状分析[J]. 大家健康:学术版, 2014, 8(7): 16-17.
[2] CHUE P, CHUE J. A review of paliperidone palmitate[J]. Expert Review of Neurotherapeutics, 2012, 12(12): 1383-1397.
[3] 蒋健昌, 李晓玲, 汤超华, 等. 棕榈酸帕利哌酮注射液与帕利哌酮缓释片对精神分裂症患者疗效和安全性的比较[J]. 广东医学, 2014, 35(9): 1414-1416.
[4] 易峰, 刘晓伟, 甄莉丽. 棕榈酸帕利哌酮注射液治疗急性精神分裂症的疗效及安全性[J]. 中国新药与临床杂志, 2013, 32(12): 988-992.
[5] 李亚辰, 李英梅, 王立娜. 棕榈酸帕利哌酮和口服帕利哌酮缓释片治疗精神分裂症患者的疗效和安全性对照研究[J]. 吉林医学, 2014, 35(15): 3217-3218.
Determination of the content and release of paliperidone palmitate long-acting suspension injection
JING Guanghui, YANG Jie, JI Xinchao, ZHONG Yue, ZHANG Yuanyuan, WANG Yanjiao*
(School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China)
ObjectivesTo establish UV methods for the determination of the content and release of paliperidone palmitate (PP) in suspension injection.MethodsThe suspension injection was prepared by wet milling. The diluent consisted of tetrahydrofuran and N,N-dimethyl formamide (V∶V=5∶95). The dissolution medium was 1 mmol·L-1hydrochloric acid solution 900 mL with 0.489% polysorbate 20. The detection wavelength was set at 280 nm.ResultsGood linear relationship (r=0.999 9) between absorbance (A) and concentration (ρ) of PP in diluent ranging from 20.0 - 60.0 mg·L-1was obtained, and the calibration curve was A=1.36×10-2ρ + 2.68×10-2(r= 0.999 9). The solution stability of PP in diluent was good, and the recoveries of low, middle and high concentrations were in the range of 98% - 102%. The intra-day and inter-day RSDs were less than 2.0%. In the dissolution medium, good linear relationship (r=0.999 9) between absorbance (A) and concentration (ρ) of PP from 6.0 - 40.0 mg·L-1was obtained, and the calibration curve was A = 1.45×10-2ρ + 9.1×10-3(r=0.999 9).ConclusionsThe solution stability of PP is good. The methods are accurate, simple, rapid and reproducible, and can be used for determination of content and release of PP in suspension injection.
pharmaceutics; long-acting suspension injection; UV; paliperidone palmitate; content determination; in vitro release
(本篇责任编辑:赵桂芝)
R 94
A
(2016)05-0175-08
10.14146/j.cnki.cjp.2016.05.004
2015-05-05
荆广会(1988-), 女(汉族), 山东威海人, 硕士研究生, E-mail jgh0410@126.com; 王艳娇(1964-), 女(汉族), 辽宁辽阳人, 副教授, 博士, 硕士生导师, 从事药物新剂型的研究与开发,Tel. 024-23986343,E-mailtangpharm@sina.com。
