(H2O)n(n=1—3)对HN(NO2)2→HONONNO2氢迁移反应的影响
2016-12-28张田雷贾子龙王炳楠
张田雷, 贾子龙, 王炳楠
(陕西理工大学 化学与环境科学学院, 陕西省催化基础与应用重点实验室, 陕西 汉中 723000)
(H2O)n(n=1—3)对HN(NO2)2→HONONNO2氢迁移反应的影响
张田雷, 贾子龙, 王炳楠
(陕西理工大学 化学与环境科学学院, 陕西省催化基础与应用重点实验室, 陕西 汉中 723000)
采用CBS-QB3方法对HN(NO2)2→HONONNO2氢迁移机理和速率常数进行理论研究,并在此基础上考虑了水簇(H2O)n(n=1—3)对该反应机理及速率常数的影响。结果表明,HN(NO2)2→HONONNO2氢迁移过程需克服33.8 kcal/mol的能垒。水簇(H2O)n(n=1—3)的加入不仅使得氢转移方式从原有反应中的直接抽氢转变为双、三、四氢原子协同转移,而且使得反应能垒降低了15.2~22.8 kcal/mol。其中,(H2O)3的参与使能垒降低22.8 kcal/mol,而(H2O)2和H2O使其分别降低22.5 kcal/mol和15.2 kcal/mol。此外,在200~1 000 K温度范围内(H2O)3参与使通道的速率常数k3比相同温度下无水参与通道的速率常数k0大了4.2×103~6.9×1024倍,说明在200~1 000 K温度范围内(H2O)3对HN(NO2)2→HONONNO2氢迁移反应所起的正催化作用最大。
HN(NO2)2; H2O; 氢迁移; 反应机理; 速率常数
在高能低特征信号固(液)体推进剂研制中,二硝酰胺铵(NH4N(NO2)2,Ammonium dinitramide, ADN)是最具发展前途的绿色高能氧化剂之一[1-6]。与传统推进氧化剂高氯酸铵(Ammonium Perchlorate, AP)、硝酸铵(AN)相比,ADN摒弃了它们在燃烧时生成大量有害气体的缺点,且具有高氧、高氮含量、无污染(不含氯)等优点。ADN推进剂可显著降低特征信号和减少对环境的污染[7],已引起全球领域的广泛兴趣,被认为是一种环境友好、性能优异的传统推进剂的替代品。ADN受热分解脱去一分子NH3,得到产物是它的共轭酸二硝酰胺酸(HN(NO2)2,Dinitramic Acid,HDN)。而HDN作为ADN的共轭酸也具有ADN的以上特点。
HDN是重要的含能材料,研究其裂解反应机理及动力学性质是化学家备受关注的研究对象,已有八十多种HDN与无机离子和有机胺形成的二硝酰胺盐被合成和研究[8-10]。在实验研究方面,因HDN(g)→N2O+HNO3反应不能通过键断裂进行,须经历分子内“O2{NONN}O”四中心重排或通过其他过渡态[11-14]。Doyle[14]通过实验提出,DN-→N2O+NO3-中存在分子内重排。虽在DN-的DFT研究中Politzer等[13]未发现分子内重排,但指出过渡态反应N(NO2)2-→NNO2-+NO2→N2O+NO3-是Ea最低通道。Politzer理论研究也支持HON(O)NNO2解离成N2O和HNO3机理。而Mebe等[15]的结果则倾向支持无酸催化下HDN发生N—NO2键断裂形成NO2。Kazakov等[16-17]认为HDN通过硝基形式HN(NO2)2分解成为N2O和HNO3。
在理论研究方面,Alavi等[7]在B3LYP/6-311G(d,p)和改进Gaussian-2水平完成了上述涉及HDN气相分解机理的反应物、产物和过渡态的几何结构、振动频率和零点能计算,给出了气相HDN最稳定结构和能量较高的4个质子转移异构体I(HN(NO2)2)、IIa、IIb、IIc和IId((HO)ONN(NO2))[18]。实验表明,水对ADN和HDN分解反应具有重要影响[19],因此,考察H2O及(H2O)2和(H2O)3对HDN分解反应的影响是很有必要的。基于此,本文采用CBS-QB3方法主要研究了H2O及(H2O)2和(H2O)3对HDN裂解反应中(HN(NO2)2)→(HONONNO2)(即Alavi等文中提及的异构体I→IIa)氢迁移过程的影响。
1 计算方法
本文所涉及的电子结构计算均使用Gaussian 09[20]程序完成。CBS是组合模型方法的一种,CBS组合模型方法是由Montgomery,Lynch等人[21-24]提出的。采用CBS-QB3方法中的B3LYP/CBSB7方法对H2O、(H2O)2和(H2O)3参与HDN裂解反应中HN(NO2)2→HONONNO2的氢迁移反应、中间体、过渡态和产物等驻点物种的几何构型进行了全参数优化,同时在相同水平下对其构型进行振动频率分析来确认所得几何构型并获得零点能。在CBS-QB3水平上各驻点物种参数分别为零点能(ZPE)、电子能(E)、相对能(△E)、焓变(△H)和吉布斯自由能变(△G)。在构建几种催化剂对HDN裂解反应中标题反应氢迁移反应势能面的基础上,使用VKLab[25]程序包,采用改进的正则变分过渡态理论(ICVT)速率常数式(1)计算标题反应各通道的速率常数kTST,
(1)
2 结果与讨论
考虑H2O及(H2O)2和(H2O)3簇溶胶对该反应机理和速率常数的影响。图1给出了HDN裂解反应中HN(NO2)2→HONONNO2的氢迁移反应通道的示意图。图2给出了在CBS-QB3水平上优化所得的反应物,中间体,过渡态和产物的几何构型,并标出对应的键长及键角值。图3是HDN中HN(NO2)2→HONONNO2氢迁移反应及其裸反应(R1)和在不同催化模型下的势能剖面图(R2、R3和R4),纵坐标为吉布斯自由能。表1列出了几种催化剂对HDN裂解反应中HN(NO2)2→HONONNO2氢迁移反应中反应物、产物、过渡态及其中间体的热力学参数(包括相对能(ΔE)、生成焓(ΔH)和吉布斯自由能(ΔG)等)。使用IM代指前中间体,IMF指后中间体,TS指过渡态,对不同催化剂H2O、(H2O)2和(H2O)3分别以IMW,IMFW,TSW表示前中间体,后中间体和过渡态,并以左上角标1—3区分表示,裸反应过渡态为TSa。
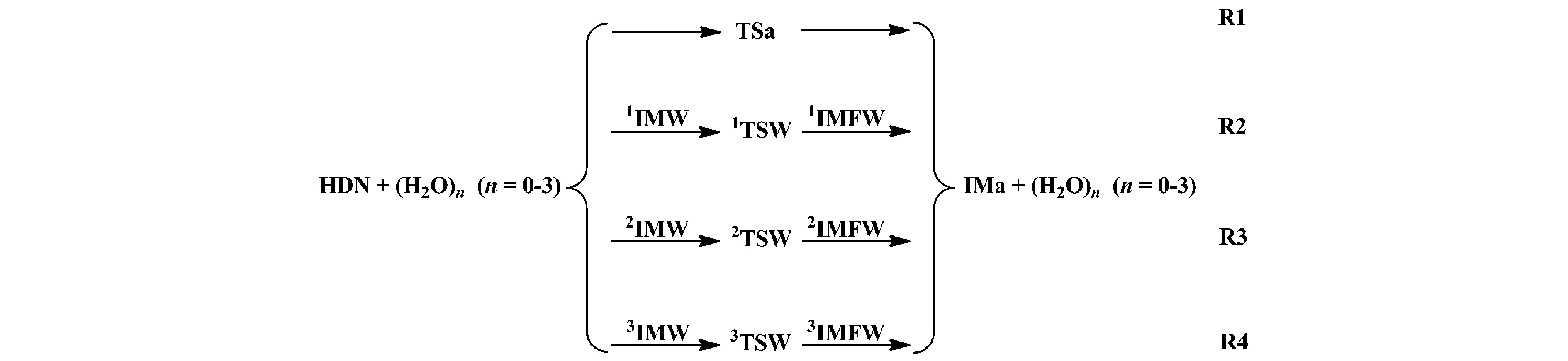
图1 无水及H2O、(H2O)2和(H2O)3簇参与下HN(NO2)2→HONONNO2氢迁移反应示意图

图2 优化所得的反应物、中间体、过渡态和产物的几何构型(键长Å,键角°)
2.1 HDN裂解反应中HN(NO2)2→HONONNO2裸反应机理
由图1、图2和图3可以看出R1反应由HDN自身克服33.8 kcal/mol的能垒形成H-N(2)-N(3)-O(4)四元环过渡态TSa,由于TSa四元环中的环张力较大使得从反应物到过渡态的能垒较高。从构型方面而言,TSa与HDN相比N(2)—H键长由原来的1.02 Å拉长为1.30 Å。在TSa中四元环中的N(2)—H和O(1)—H键的键长均为1.30 Å,但由于O(1)—H中氢键的作用较N(2)—H中氢键的作用力大,故此N(2)—H键断裂,O(1)—H键形成,生成产物IMa。IMa与HDN相比其O(1)—H的键长为0.97 Å。在此过程中反应需要吸收3.7 kcal/mol的能量,表明此反应在热力学上不易发生。
2.2 催化剂条件下HDN裂解反应中HN(NO2)2→HONONNO2氢迁移反应机理
H2O、(H2O)2和(H2O)3等催化剂参与反应的过程类似于HN(NO2)2→HONONNO2氢迁移裸反应过程,但其氢转移方式不同于裸反应中的直接抽氢,在有催化剂参与的反应中氢转移方式分别为双、三和四氢原子转移。加入水簇时,由于反应过程中其经历的过渡态张力较小,簇合物的质子可以帮助氢原子迁移,使得氢迁移过程更加顺利的进行[26]。
2.2.1 H2O催化条件下HN(NO2)2→HONONNO2氢迁移反应机理
H2O作为催化剂的氢转移方式类似于HN(NO2)2→HONONNO2反应过程的氢迁移,但其不同于裸反应的直接抽氢方式,H2O催化条件下标题反应氢迁移反应是双氢原子转移。由图1和图3可以看出,对R2反应而言,HN(NO2)2与H2O先形成六元环的前产物中间体1IMW。由能量角度而言,HN(NO2)2与H2O形成的前反应物中间体1IMW稳定化能相比HN(NO2)2的能量高了0.8 kcal/mol,由前反应物中间体1IMW自身克服了17.8 kcal/mol的能垒形成H(1)-O(1)-N(3)-N(2)-H-O(5)六元环过渡态1TSW。从几何构型角度而言,1TSW与TSa相比,由于1TSW中形成的是六元环过渡态,其六元环的分子间双氢原子转移比TSa中四元环的分子内氢原子转移更加容易。其次由于1TSW中的N—H键的键长为1.47 Å,比TSa中的N—H键的键长拉长了0.17 Å。因此,氢原子更容易迁移。所以R2反应的相对能垒比R1反应的相对能垒降低了15.2 kcal/mol。在1TSW中六元环中的N(2)—H和O—H(1)键断裂,O(1)—H(1)键形成,生成后产物中间体1IMFW,其后产物中间体1IMFW的相对能量为4.2 kcal/mol。后产物中间体1IMFW中的H2O离去,形成产物HONONNO2,但产物HONONNO2较HN(NO2)2不稳定。
比较R1与R2,表明H2O作为催化剂,对HN(NO2)2裂解反应中标题反应氢迁移反应具有明显的正催化作用,有效的降低了标题反应的能垒,使得H2O催化条件下HONONNO2裂解反应中标题反应氢迁移反应比其裸反应在动力学上更容易发生。

图3 HN(NO2)2→HONONNO2氢迁移裸反应及不同催化剂参与下的势能剖面图

种类ΔE/(kcal·mol-1)ΔH/(kcal·mol-1)ΔG/(kcal·mol-1)HN(NO2)20.00.00.0TSa33.533.533.8IMa4.44.43.7HN(NO2)2+H2O0.00.00.01IMW-7.1-7.70.81TSW7.77.218.61IMFW-4.4-5.04.2HN(NO2)2+(H2O)20.00.00.02IMW-11.2-11.2-1.12TSW-2.4-3.011.32IMFW-10.8-11.31.1HN(NO2)2+(H2O)30.00.00.03IMW-12.0-12.6-1.63TSW1.10.511.03IMFW-10.8-11.40.8
2.2.2 (H2O)2催化条件下HN(NO2)2→HONONNO2氢迁移反应机理
(H2O)2作为催化剂的氢转移方式类似于H2O催化条件下HN(NO2)2→HONONNO2氢迁移反应方式,但其不同于H2O催化条件下标题反应的双氢原子转移方式,(H2O)2催化条件下标题反应的氢迁移反应机理经历了三氢原子转移方式。由图2和图3可以看出,对R3反应而言,HN(NO2)2与H2O先形成八元环的前产物中间体2IMW。从能量角度而言,HN(NO2)2与(H2O)2形成的前反应物中间体2IMW稳定化能相比HN(NO2)2的能量降低了1.1 kcal/mol,由前反应物中间体2IMW出发,反应克服了12.4 kcal/mol的能垒形成H(1)-O(5)-H-N(2)-N(3)-O(1)-H(2)-O(6)八元环过渡态2TSW。从能量角度而言,R3反应的相对能垒比R2反应的相对能垒降低了7.3 kcal/mol,表明(H2O)2作为催化剂,对标题反应氢迁移反应具有明显的正催化作用,有效降低了标题反应的活化能,使反应能垒相比H2O的加入又降低7.3 kcal/mol。
2.2.3 (H2O)3催化条件下HN(NO2)2→HONONNO2氢迁移反应机理
(H2O)3作为催化剂的反应方式类似于H2O和(H2O)2催化条件下HN(NO2)2→HONONNO2氢迁移反应机理,但不同于H2O和(H2O)2催化条件下标题反应中所经历的双氢原子转移方式和三氢原子转移方式,(H2O)3催化条件下标题氢迁移反应经历了四氢原子转移过程。与H2O和(H2O)2催化反应类似,由图1和图3知,R4反应同样经历了类似的HN(NO2)2与(H2O)3首先形成十元环的前产物中间体3IMW。由能量方面而言,3IMW的能量相比HN(NO2)2降低了1.6 kcal/mol。由3IMW出发,R4反应克服了12.6 kcal/mol的能垒形成H-O(5)-H(1)-O(6)-H(2)-O(7)-H(3)-O(1)-N(3)-N(2)十元环过渡态3TSW。之后,3TSW中的N(2)—H和O(7)—H(3)键断裂,O(1)—H(3)键形成,形成后中间体3IMFW。由于3IMFW中氢键数目较多,故其相对能量比1IMFW和2IMFW的能量分别降低了3.4和0.3 kcal/mol。与HN(NO2)2→HONONNO2裸反应相比,H2O、(H2O)2、(H2O)3催化HN(NO2)2→HONONNO2氢迁移反应均具有正催化作用,均降低了标题反应的活化能。从催化效果来看,(H2O)3参与HN(NO2)2→HONONNO2反应使得反应能垒降低了22.8 kcal/mol,比(H2O)2和H2O参与标题反应的能垒分别降低了7.6和0.3 kcal/mol。因此,从能垒降低角度而言,(H2O)3比(H2O)2和H2O作为催化剂的效果更合适。
2.3 反应速率常数计算
在上述有、无催化剂(H2O、(H2O)2、(H2O)3)参与HN(NO2)2→HONONNO2反应机理的基础上,进一步研究催化剂(H2O、(H2O)2、(H2O)3)在200~1 000 K温度范围内对HN(NO2)2→HONONNO2反应的影响。表2给出了各条路径的速率常数信息,图4给出了表观速率常数随温度的变化曲线。
表2 有、无催化剂(H2O、(H2O)2、(H2O)3)参与HN(NO2)2→HONONNO2氢迁移反应各路径的速率常数信息
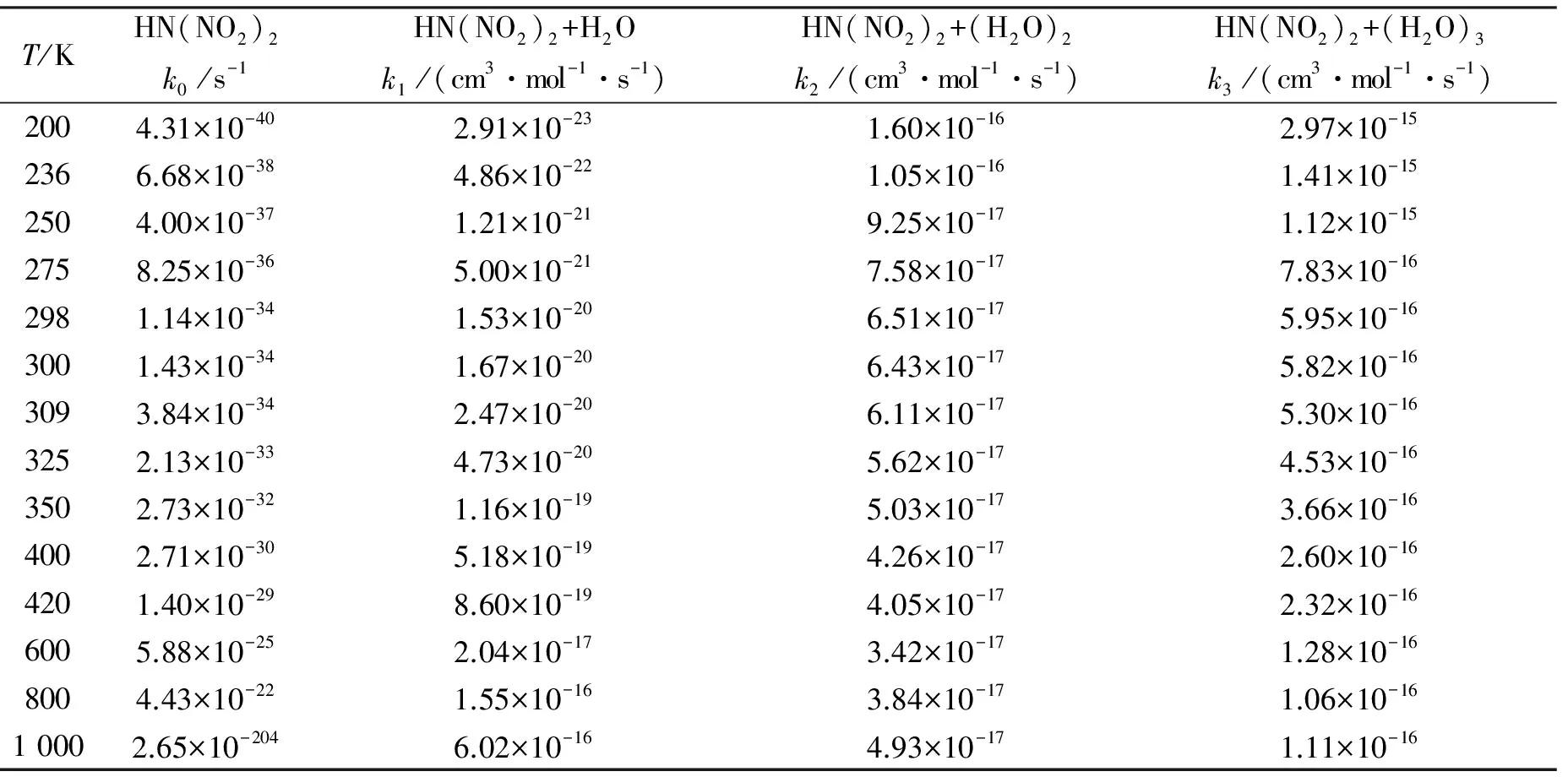
T/KHN(NO2)2k0/s-1HN(NO2)2+H2Ok1/(cm3·mol-1·s-1)HN(NO2)2+(H2O)2k2/(cm3·mol-1·s-1)HN(NO2)2+(H2O)3k3/(cm3·mol-1·s-1)2004.31×10-402.91×10-231.60×10-162.97×10-152366.68×10-384.86×10-221.05×10-161.41×10-152504.00×10-371.21×10-219.25×10-171.12×10-152758.25×10-365.00×10-217.58×10-177.83×10-162981.14×10-341.53×10-206.51×10-175.95×10-163001.43×10-341.67×10-206.43×10-175.82×10-163093.84×10-342.47×10-206.11×10-175.30×10-163252.13×10-334.73×10-205.62×10-174.53×10-163502.73×10-321.16×10-195.03×10-173.66×10-164002.71×10-305.18×10-194.26×10-172.60×10-164201.40×10-298.60×10-194.05×10-172.32×10-166005.88×10-252.04×10-173.42×10-171.28×10-168004.43×10-221.55×10-163.84×10-171.06×10-1610002.65×10-2046.02×10-164.93×10-171.11×10-16
注:k0为HN(NO2)2→(HONONNO2)的速率常数,k1为HN(NO2)2+H2O→(HONONNO2)+H2O的速率常数,k2为HN(NO2)2+(H2O)2→(HONONNO2)+(H2O)2的速率常数,k3为HN(NO2)2+(H2O)3→(HONONNO2)+(H2O)3的速率常数。

图4 4个通道的表观速率常数随温度的变化曲线
在过渡态TS和产物P(products)之前,有无催化剂(H2O、(H2O)2、(H2O)3)参与HN(NO2)2→HONONNO2反应涉及到的反应路径首先形成一个无能垒的中间体IM(见图3),
(2)
假设如(2)式所示中间体IM(Intermediate)与反应物处于平衡状态,根据稳态条件,速率常数k为
(3)
如果k2≪k-1,(3)式所示的速率常数k则可以表示为
(4)
其中,Keq和k2可通过(5)式和VKLab程序包获得。
(5)
由表2所示的HN(NO2)2→HONONNO2裸反应通道path1的速率常数k0可知,在298 K,通道path1的速率常数k0为1.14×10-34s-1。正如上述机理部分所述,H2O、(H2O)2、(H2O)3均对HN(NO2)2→HONONNO2氢迁移反应起正催化作用且降低了标题反应的生成能垒。对于H2O、(H2O)2、(H2O)3参与通道path2、path3和path4,由表2可知,在200~1 000 K温度范围内通道path2、path3和path4的速率常数k1、k2和k3分别为比相同温度下裸反应通道path1的速率常数k0大了2.27×104~6.75×1016、1.86×103~3.71×1023和4.19×103~6.89×1024倍,说明H2O、(H2O)2、(H2O)3参与HN(NO2)2→HONONNO2反应均有利于该反应的发生。此外,在计算温度范围内通道path4的速率常数k3为2.97×10-15~1.11×10-16cm3·mol-1·s-1,比相同温度下H2O、(H2O)2参与通道path2和path3的速率常数k1和k2大了1.84×10-1~1.02×108和2.25~1.86×101倍,说明经由通道path4,(H2O)3对HN(NO2)2→HONONNO2氢迁移反应的发生起显著的正催化作用。
由图4知,在200~1 000 K温度范围内,在lnk随着温度的增大而增大,呈正温度系数效应。在低温段范围内,(H2O)3催化HN(NO2)2→HONONNO2氢迁移反应表观速率常数与裸反应相比没有其在高温段更加优势,在高温段范围内,(H2O)3催化HN(NO2)2→HONONNO2氢迁移反应表观速率常数与裸反应相比优势较低温段明显减小,这是由于裸反应在温度升高时速率常数不断增大,而(H2O)3催化HN(NO2)2→HONONNO2氢迁移反应表观速率常数在高温和低温段没有较大变化的结果。不同于裸反应与(H2O)3,(H2O)2催化HN(NO2)2→HONONNO2氢迁移反应表观速率常数随温度变化无论在高温段还是低温段都与(H2O)3相近,H2O则在低温段介于(H2O)3和(H2O)2之间,其在高温段不可思议的高于(H2O)3和(H2O)2的表观速率常数。
3 结 论
本文采用CBS-QB3方法对HN(NO2)2→HONONNO2氢迁移机理和速率常数进行了研究。并在此基础上考虑了H2O、(H2O)2和(H2O)3对该反应机理及速率常数的影响,得到主要结论如下:
(1)HN(NO2)2→HONONNO2氢迁移裸反应经四元环过渡态TSa越过33.8 kcal/mol的能垒生成产物HONONNO2,在200~1 000 K的温度范围内,HN(NO2)2→HONONNO2氢迁移反应的速率为4.31×10-40~2.65×10-20s-1,表明该反应在动力学上不易发生。
(2)H2O、(H2O)2和(H2O)3的加入并没有改变标题反应的产物,却使得氢转移方式由原有裸反应中的直接抽氢变为双、三、四氢原子转移。从能垒改变角度而言,H2O、(H2O)2和(H2O)3的加入使标题反应的能垒降低了16.0~21.4 kcal/mol,其中(H2O)3参与反应的能垒降低值仅与(H2O)2相差0.2 kcal/mol。
(3)在200~1 000 K温度范围内,(H2O)3作为催化剂对标题反应催化的速率常数最大,为2.97×10-15~1.1×10-16cm3·mol-1·s-1。故此,主反应通道为(H2O)3作为催化剂的反应通道,即path4。在H2O及(H2O)2和(H2O)3几种催化剂中(H2O)3的催化效果最为明显。
[1] BOTTARO J C, PENWELL P E, SCHMITT R J. 1, 1, 3, 3-Tetraoxo-1, 2, 3-Triazapropene anion, a new oxy anion of nitrogen: the dinitramide anion and its salts[J]. Journal of the American Chemical Society, 1997, 119(40): 9405-9410.
[2] VENKATACHALAM S, SANTHOSH G, NINAN K N. An overview on the synthetic routes and properties of ammonium dinitramide (ADN) and other dinitramideSalts[J]. Propellants Explosives Pyrotechnics, 2004, 29(3): 178-187.
[3] YANG R, THAKRE P, YANG V. Thermal decomposition and combustion of ammonium dinitramide (Review)[J]. Combustion Explosion and Shock Waves, 2005, 41(6): 657-679.
[4] BADGUJAR D M, TALAWAR M B, ASTHANA S N, et al. Advances in science and technology of modern energetic materials: an overview[J]. Journal of Hazardous Materials, 2008, 151(2): 289-305.
[5] TALAWAR M B, SIVABALAN R, ANNIYAPPAN M, et al. Emerging trends in advanced high energy materials[J]. Combustion Explosion and Shock Waves,2007, 43(1): 62-72.
[6] BORMAN S. Alzheimers-disease-free-radical reactions may play key role[J]. Chemical Engineering News, 1994, 72(16):4-5.
[7] ALAVI M, LEIDNER D E. Review: knowledge management and knowledge management systems: conceptual foundations and research issues[J]. MIS quarterly, 2001, 25(1): 107-136.
[8] VENKATACHALAM S, SANTHOSH G, NINAN K N. An Overview on the Synthetic Routes and Properties of Ammonium Dinitramide (ADN) and other Dinitramide Salts[J]. Propellants Explosives Pyrotechnics, 2004, 29(3):178-187.
[9] SINGH R P, VERMA R D, MESHRI D T, et al. Energetic nitrogen rich salts and ionic liquids[J]. Angewandte Chemie International Edition, 2006, 45(22): 3584-3601.
[10] TANBUG R, KIRSCHBAUM K, PINKERTON A A. Energetic materials: The preparation and structural characterization of melaminium dinitramide and melaminium nitrate[J]. Journal of Chemical Crystallography, 1999, 29 (1): 45-55.
[11] RUSSELL T P, PIERMARINI G J, BLOCK S, et al. Pressure, temperature reaction phase diagram for ammonium dinitramide[J]. The Journal of Physical Chemistry, 1996, 100(8): 3248-3251.
[12] OXLEY J C, SMITH J L, ZHENG W, et al. Thermal decomposition studies on ammonium dinitramide (ADN) and 15N and 2H Isotopomers[J]. The Journal of Physical Chemistry A, 1997, 101(31): 5646-5652.
[13] POLITZER P, SEMINARIO J M, CONCHA M C, et al. Density functional study of the structure and some decomposition reactions of the dinitramide anion N(NO2)2[J]. Journal of Molecular Structure, 1993, 287(106): 235-240.
[14] DOYLE R J. Sputtered ammonium dinitramide: tandem mass spectrometry of a new ionic nitramine[J]. Organic Mass Spectrometry, 1993, 28(2): 83-91.
[15] MEBEL A M, LIN M C, MOROKUMA K, et al. Theoretical study of the gas-phase structure, thermochemistry, and decomposition mechanisms of NH4NO2and NH4N(NO2)2[J]. The Journal of Physical Chemistry, 1995, 99(18): 6842-6848.
[16] KAZAKOV A I, RUBTSOV Y I, MANELIS G B, et al. Kinetics of the thermal decomposition of dinitramide. 2. kinetics of the reactions of dinitramide with decomposition products and other components of a solution[J]. Russian Chemical Bulletin, 1998, 47(1): 39-45.
[17] KAZAKOV A I, RUBTSOV Y I, MANELIS G B, et al. Kinetics of thermal decomposition of dinitramide[J]. Russian Chemical Bulletin, 1997, 46(12): 2015-2020.
[18] RAHM M. BRINCK T.Dinitraminic acid ( HDN) isomerization and self-decomposition revisited[J]. Chemical Physics,2008,348(1-3):53-60.
[19] 王志银,许琼,张田雷,等.二硝酰胺铵热分解机理的理论研究进展[J].含能材料,2015,23(9):831-841.
[20] FRISCH M J, TRUCKS G W, POPLE J A, et al. Gaussian 09, revision A. 01[CP]. Gaussian inc: pittsburgh, pa, 2009.
[21] MONTGOMERY J A, FRISCH M J, OCHTERSKI J W, et al. A complete basis set model chemistry. VI. use of density functional geometries and frequencies[J]. The Journal of Chemical Physics, 1999, 110(6): 2822-2827.
[22] MONTGOMERY J A, OCHTERSK J W, PETERSSON G A. A complete basis set model chemistry. IV. an improved atomic pair natural orbital method[J]. Journal of Chemical Physics, 1994, 101(7): 5900-5909.
[23] LYNCH B J, ZHAO Y, TRUHLAR D G. Effectiveness of diffuse basis functions for calculating relative energies by density functional theory[J]. The Journal of Physical Chemistry, 2003, 107(9): 1384-1388.
[24] PETERSSON G A, TENSFELDT T G, MONTGOMERY J A. A complete basis set model chemistry. III. The complete basis set quadratic configuration interaction family of methods[J]. The Journal of Chemical Physics, 1991, 94(9): 6091-6101.
[25] ZHANG S, TRUONG T N. VKLab version 1.0[CP]. Salt Lake City: University of Utah,2001.
[26] 朱艳艳,魏东辉,张文静,等.几种常见氢迁移反应的理论新见解[J].大学化学,2014,29(4):52-57.
[责任编辑:张存凤]
Effects of (H2O)n(n= 1—3) on hydrogen transfer process
of HN(NO2)2→HONONNO2
ZHANG Tian-lei, JIA Zi-long, WANG Bing-nan
(School of Chemistry and Environment Science, Shaanxi Key Laboratory of Catalysis,Shaanxi Sci-Tech University, Hanzhong 723000,China)
The effect of (H2O)n(n= 1—3) on the hydrogen abstraction reaction of HN(NO2)2→HONONNO2has been investigated by employing CBS-QB3 method and conventional transition state theory with wigner tunneling correction. The reaction without catalysts overcomes the barrier height of 33.8 kcal/mol to form HONONNO2. (H2O)n(n= 1—3) does not change the product of the reaction, but the direct hydrogen abstraction mechanism changes to double, three, and four hydrogen transfer mechanism. Compared with the naked reaction, the barrier height of the hydrogen abstraction reaction with (H2O)n(n= 1—3) is lower by 15.2 ~ 22.8 kcal/mol. Moreover, the barrier height of (H2O)3assisted reaction is the lowest. Besides, the rate constants without and with catalyst show that the rate constant of (H2O)3-assisted channel is predicted to be faster by 4.2×103~ 6.9 × 1024times than that of the naked hydrogen transfer reaction at 200 ~ 1 000 K, showing that, in temperature range 200 ~ 1 000 K, the (H2O)3plays a positive catalytic role for the reaction of HN(NO2)2→HONONNO2.
HN(NO2)2; H2O; hydrogen transfer; reaction mechanism; rate constant
1673-2944(2016)06-0057-08
2016-07-08
2016-09-18
国家自然科学基金资助项目(21603132);陕西省教育厅科研计划项目(14JK1154)
张田雷(1982—),男,山西省朔州市人,陕西理工大学讲师,博士,主要研究方向为理论计算化学。
O643.1
A
