HBF4催化末端炔烃水化成甲基酮的合成研究*
2016-12-21叶东鼐傅叶娟王青豪
叶东鼐,傅叶娟,王青豪,叶 敏
(赣南师范大学 化学化工学院,江西 赣州 341000)
·有机药物化学·
HBF4催化末端炔烃水化成甲基酮的合成研究*
叶东鼐,傅叶娟,王青豪,叶 敏†
(赣南师范大学 化学化工学院,江西 赣州 341000)
炔烃水化成酮是有机合成中最基本的转化.本论文以四氟硼酸为催化剂,炔烃为反应底物, 三氟乙醇(TFE)作溶剂,高效水化合成一系列甲基酮类化合物.考察了不同反应条件,拓展了反应的底物,并提出了反应的可能机理.
甲基酮;末端炔;水化;催化
酮类化合物是最重要的合成原料之一,将炔烃化合物进行Markovnikov类型的水化反应合成酮是有机合成中最基本的转化,该反应具有100%的原子利用率[1].一般认为在酸催化下,炔烃直接水合比较困难,但在硫酸汞的硫酸溶液催化下,炔烃较易与水发生加成反应,称为Kucherov反应(图1, a)[2].反应中所用汞盐有剧毒,因此很早已开始非汞催化剂的研究,最近取得了比较大的进展.所用催化剂主要有钌[3]、铑[4]、钯[5]、铂[6]、金[7]、银[8]、铱[9]、钴[10]、铁[11]、锡-钨[12]等过渡金属催化.另外Bronsted酸催化的炔烃水化反应仅有少数文献报道[13-16].尽管如此,已经报道的这些方法仍存在一些缺点:比如需要高温反应、使用到昂贵的金属、官能团兼容性差以及大大过量的酸作为添加剂(图1, b).因此继续探寻操作简单、官能团兼容性好、无过渡金属的方法合成酮类化合物仍是有机合成的热点.

图1 文献中有关炔烃水化成酮的介绍
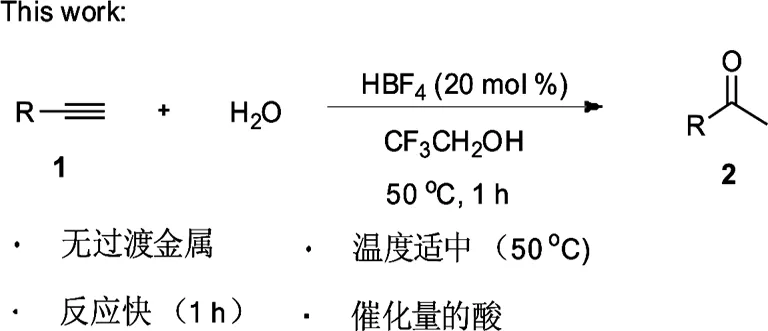
图2 四氟硼酸催化末端炔烃水化成甲基酮
1 实验部分
1.1 仪器与试剂
Bruker AM-400 FT-NMR核磁共振仪(TMS为内标,CDCl3为溶剂);岛津GCMS-QP5050A 质谱仪;反应试剂均为国产市售分析纯.
1.2 HBF4催化末端炔烃水化合成甲基酮的实验操作
于25 mL螺旋刻度试管中分别加入0.2 mmol炔烃1、四氟硼酸 (20 mol %)(四氟硼酸为40%的水溶液)和1 mL 三氟乙醇,50 ℃反应1 h.反应结束后,加入10 mL水,乙酸乙酯(3×5 mL)萃取,合并有机层并用无水硫酸镁进行干燥.浓缩,去溶剂.所得余留物经柱色谱分离,得到相应的酮化合物2.
1.3 产物结构分析
苯乙酮(2a):1H NMR(400 MHz, CDCl3):δ=7.94-7.96(m,2H), 7.53-7.58(m,1H),7.44-7.47 (m,2H), 2.60 (s,3H).13C NMR (100 MHz,CDCl3):δ=198.1, 137.1, 133.1, 128.5, 128.3, 26.6. MS(EI)m/z: 120, 105, 71, 51.
3-甲基苯乙酮(2b):1H NMR(400 MHz,CDCl3):δ=7.75(d,J=10.8 Hz,2H),7.33(t,J=8.0 Hz, 2H),2.58(s,3H),2.40(s,3H).13C NMR(100 MHz,CDCl3):δ= 198.4, 138.3, 137.4, 133.8, 128.7, 128.4, 125.5, 26.6, 21.3. MS(EI)m/z: 134, 119, 91, 65, 51.
4-叔丁基苯乙酮(2c):1H NMR(400 MHz,CDCl3):δ=7.91 (t,J=3.0 Hz,1H),7.89(t,J= 3.0 Hz,1H),7.49(t,J= 3.0 Hz,1H),7.46(t,J= 3.0 Hz, 1H),2.58(s, 3H),1.34(s,9H).13C NMR(100 MHz,CDCl3):δ=197.9, 156.8, 134.6, 128.2, 125.5, 35.1, 31.0, 26.5. MS(EI)m/z: 176, 161, 133, 118, 91, 77.
4-苯基苯乙酮(2d):1H NMR(400 MHz,CDCl3):δ=8.02-8.05(m,2H),7.67-7.70(m,2H),7.61-7.64(m,2H),7.45-7.49(m,2H),7.38-7.42(m,1H),2.64(s,3H).13C NMR(100 MHz,CDCl3):δ=197.8, 145.7, 139.8, 135.8, 128.9, 128.9, 128.2, 127.2, 127.2, 26.6. MS (EI) m/z: 196, 181, 152, 76, 43.
4-甲氧基苯乙酮(2e):1H NMR (400 MHz, CDCl3):δ=7.91 (d,J= 8.8 Hz, 2H),6.90 (d,J= 8.8 Hz, 2H), 3.84 (s, 3H), 2.52 (s, 3H).13C NMR (100 MHz, CDCl3):δ= 196.7, 163.4, 130.5, 130.3, 113.6, 55.4, 26.3. MS (EI) m/z: 150, 135, 107, 92, 77, 63.
2-甲氧基苯乙酮(2f):1H NMR(400 MHz,CDCl3):δ=7.69-7.72(m,1H),7.41(m,1H),6.93-6.98(m,2H),3.88(s,3H),2.59(s,3H).13C NMR(100 MHz,CDCl3):δ= 199.8, 158.9, 133.6, 130.3, 128.2, 120.5, 111.6, 55.4, 31.8. MS(EI)m/z: 150, 135, 105, 92, 77, 63.
4-氟苯乙酮(2g):1H NMR(400 MHz,CDCl3):δ=7.92-7.95 (m,2H),7.06-7.10(m,2H),2.54(s,3H).13C NMR(100 MHz,CDCl3):δ= 196.4, 166.9, 164.4, 133.6, 133.5, 130.9, 130.8, 115.7, 115.4, 26.4. MS(EI)m/z: 138, 123, 95, 75.
4-氯苯乙酮(2h):1H NMR(400 MHz,CDCl3):δ=7.82(d,J=8.4 Hz,2H),7.36(d,J= 8.4 Hz,2H),2.52(s,3H).13C NMR(100 MHz,CDCl3):δ= 196.7, 139.4, 135.4, 129.6, 128.8, 26.4. MS(EI)m/z: 154, 139, 111, 74.
4-溴苯乙酮(2i):1H NMR(400 MHz,CDCl3):δ=7.80(d,J=8.4 Hz,2H),7.58(d,J=8.4 Hz,2H),2.56(s,3H).13C NMR(100 MHz,CDCl3):δ= 196.9, 135.8, 131.8, 129.8, 128.2, 26.5. MS(EI)m/z: 198, 183, 155, 104, 75.
4-三氟甲基苯乙酮(2j):1H NMR(400 MHz,CDCl3):δ=8.06(d,J=8.0 Hz,2H),7.73(d,J=8.0 Hz,2H),2.65(s,3H).13C NMR(100 MHz,CDCl3):δ= 196.9, 139.6, 134.2, 134.2, 128.6, 125.7, 125.7, 125.6, 125.6, 124.9, 26.7. MS(EI)m/z: 188, 173, 145, 125, 95, 75, 50.
2 结果与讨论
2.1 反应条件的筛选
我们以0.2 mmol苯乙炔作为反应原料,加入20 mol % 的酸和1 mL溶剂,室温下反应为模型反应,进行了条件的优化.我们首先以三氟乙醇为反应溶剂,尝试了不同的酸.如表1所示,醋酸和三氟醋酸作为催化剂不能得到对应的产物,而四氟硼酸能有效催化该反应的进行,得到70%的收率.其次我们以四氟硼酸为反应的催化剂,考察了不同的溶剂对该反应的影响.发现反应的溶剂对反应的顺利进行非常的重要,除了以三氟乙醇为溶剂能得到对应的产物,其它的溶剂,极性溶剂、非极性溶剂、质子性溶剂和非质子性溶剂都不能得到对应的产物.唯一的例外是三氟乙醇的类似物HFIP作为溶剂能得到50%收率的产物.最后我们考察了反应的温度,将温度提高到50度反应,能得到近似定量收率的产物.因此,综合以上因素,该反应的最佳条件为:四氟硼酸为催化剂,三氟乙醇为溶剂,50 ℃反应1 h.
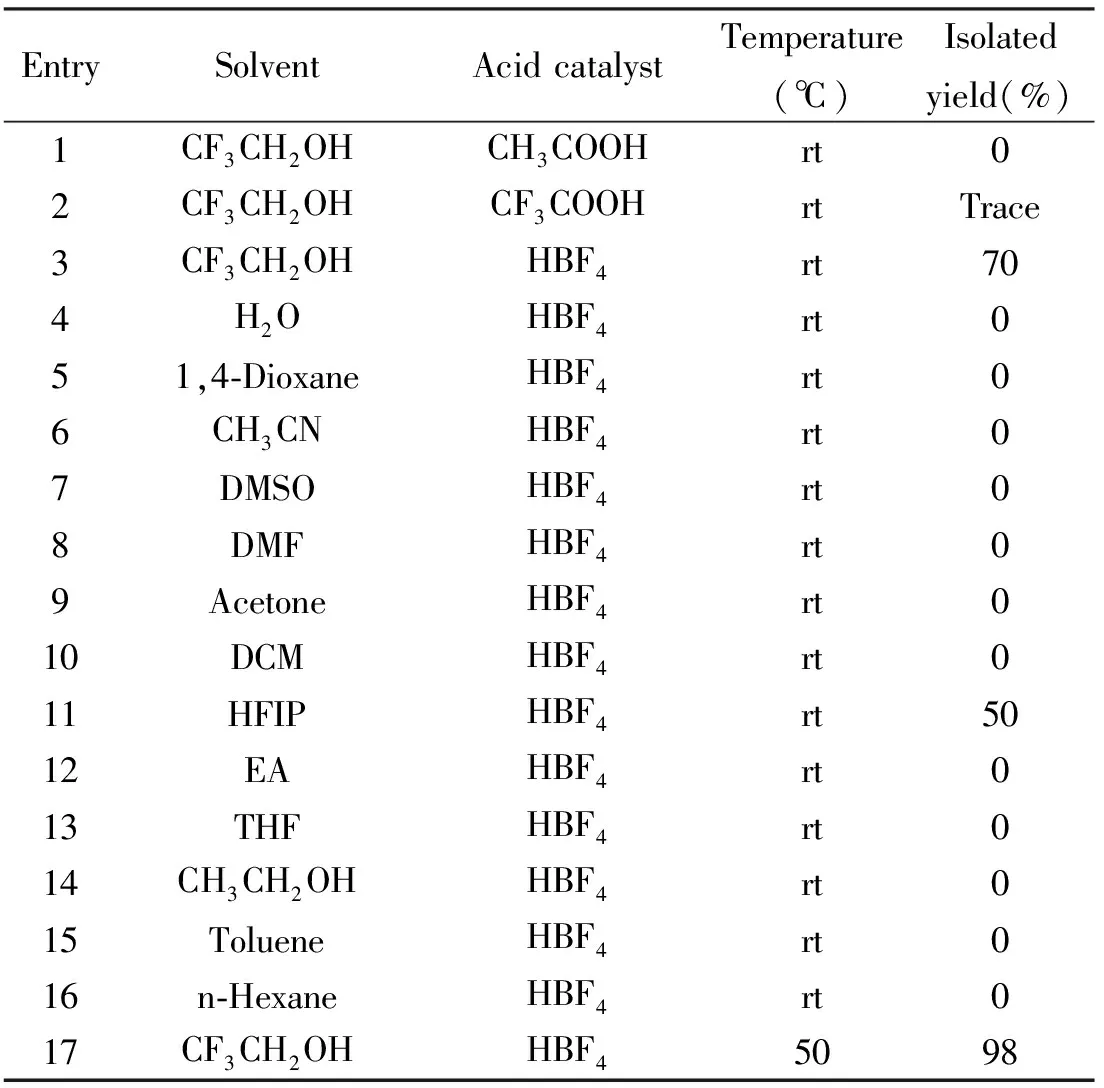
表1 反应条件的优化
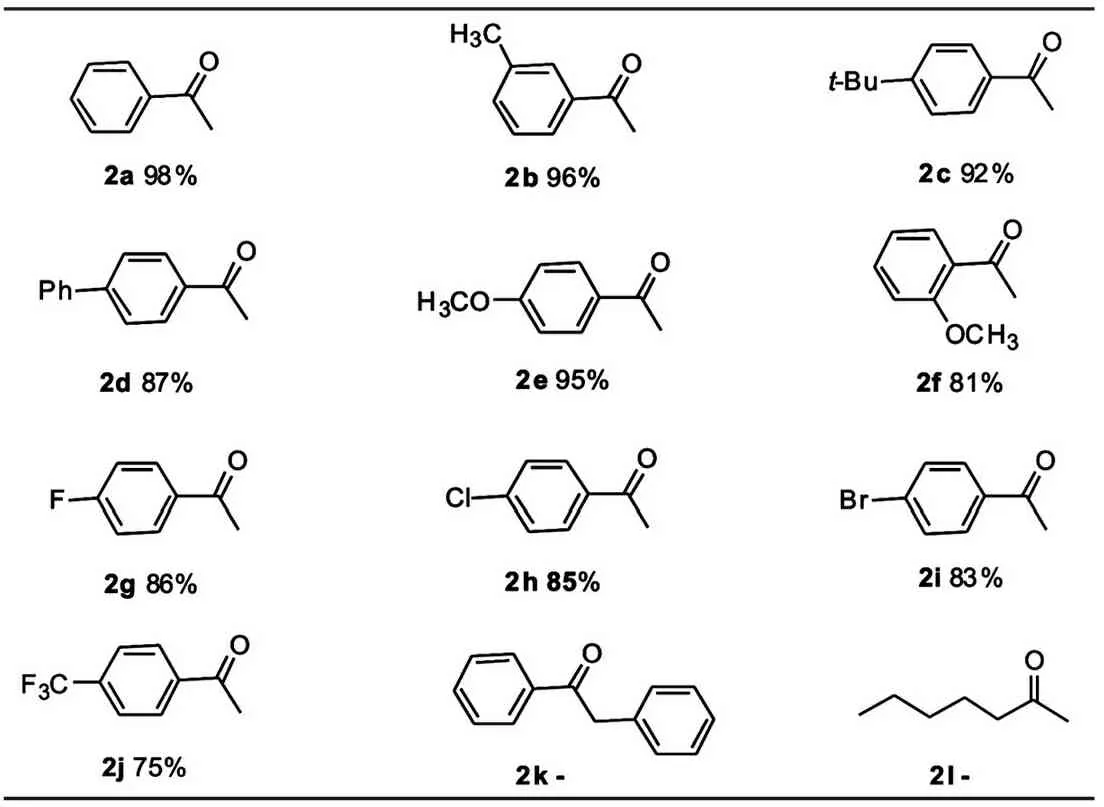
表2 酸催化炔烃水化成酮的底物拓展

图3 四氟硼酸催化末端炔烃合成甲基酮的反应机理
2.2 反应底物拓展
在最优化的反应条件下,我们对反应的底物进行了拓展.如表2所示,选取了有代表性的炔烃进行了测试.总体而言,苯环上连有给电子基团和吸电子基团的底物都能得到对应的产物,但给电子基团更有利于反应的进行,这可能是由于当苯环上有给电子基团时,更能稳定中间体烯基碳正离子,有利于反应的进行.当苯环对位有体积比较大的基团如叔丁基时,收率没有受到明显的影响.当乙炔基的邻位有取代基时,由于空间位阻的影响,收率有一定的影响,但仍能得到较高的收率.对于苯环上连接有吸电子基时,收率有一定的降低,尤其是当有强拉电子基团,收率降低比较明显.值得注意的是含有卤素原子的底物能兼容该反应体系,这些产物能够在过渡金属的催化下,方便的实现进一步的转化.在优化条件下,我们也尝试了内炔烃,发现该底物没有反应,这样有助于实现内炔和末端炔烃的官能团选择性.此外,脂肪末端炔烃也不能发生反应,这可能是由于芳香炔烃能形成稳定的中间体,而脂肪炔烃相应的中间体不如芳香炔烃稳定.
2.3 可能的反应机理
基于之前的报道和上述的实验结果,提出了可能的反应机理,如图3所示.首先,在三氟乙醇溶剂中,末端炔烃与酸进行亲电加成得到烯基正离子中间体A, 该过程遵从Markovnikov规则.由于三氟甲基的强吸电子作用导致三氟乙醇不能与中间体A作用,烯基正离子与水作用得到烯醇中间体B, 最后烯醇互变成甲基酮产物2.值得注意的是,该体系的另外一个阴离子即四氟硼酸根,同样不是一个好的亲核试剂,难与烯基正离子作用,正是由于这些原因,该转化反应速度较快.
3 结论
本文介绍了一种以三氟乙醇为溶剂,酸催化炔烃水化成酮的方法.避免了以往反应体系常用过渡金属作为催化剂、酸为溶剂的合成体系.该反应温度适中,反应速度较快.此外该反应具有较好的官能团兼容性,得到较高的分离收率.本催化体系将末端炔烃转化成甲基酮的方法的实际应用和机理探讨还在继续研究中.
[1] Hintermann L, Labonne A. Catalytic hydration of alkynes and its application in synthesis[J].Synthesis 2007,8:8933-8935.
[2] Budde W L, Dessy R E. The homogeneously catalyzed hydration of acetylenes by mercuric perchlorate-perchloric acid: Evidence for a bis-(acetylene)-mercuric ion complex as an intermediate[J].J. Am. Chem. Soc. 1963,85:3964-3970.
[3] Imi K, Imai K, Utimoto K. Regioselective hydration of alkynones by palladium catalysis[J].Tetrahedron Lett. 1987,28:3127-3130.
[4] Wu X-F, Bezier D, Darcel C. Development of the first iron chloride-catalyzed hydration of terminal alkynes[J].Adv. Synth. Catal. 2009,351:367-370.
[5] Tachinami T, Nishimura T, Ushimaru R, et al. Hydration of terminal alkynes catalyzed by water-soluble cobalt porphyrin complexes[J].J. Am. Chem. Soc. 2013,135:50-53.
[6] Chen Z-W, Ye D-N, Qian Y-P, et al. Highly efficient AgBF4-catalyzed synthesis of methyl ketones from terminal alkynes[J].Tetrahedron 2013,69:6116-6120.
[7] Xu Y, Hu X, Shao J, et al. Hydration of alkynes at room temperature catalyzed by gold isocyanide compounds[J].Green Chem. 2015,17:532-537.
[8] Labonne A, Kribber T, Hintermann L. Highly active in situ catalysts for anti-Markovnikov hydration of terminal alkynes[J].Org. Lett. 2006,8:5853-5856.
[9] Kanemitsu H, Uehara K, Fukuzumi S, et al. Isolation and crystal structures of both enol and keto tautomer intermediates in a hydration of an alkyne-carboxylic aicd ester catalyzed by iridium complexes in water[J].J. Am. Chem. Soc. 2008,130:17141-17147.
[10] James B R, Rempel G L. Hydration of acetylenes catalyzed by rhodium chloride complexs[J].J. Am. Chem. Soc. 1969,91:863-865.
[11] Baidossi W, Lahav M, Blum J. Hydration of alkynes by a PtCl4-CO catalyst[J].J Org. Chem. 1997,62:669-672.
[12] Jin X, Oishi T, Yamaguchi K, et al. Heterogeneously catalyzed efficient hydration of alkynes to ketones by tin-tungsten mixed oxides[J].Chem. Eur. J. 2011,17:1261-1267.
[13] Wong W-L, Ho K-P, Lee L Y S, et al. Sulfuric acid-catalyzed conversion of alkynes to ketones in an ionic liquid medium under mild reaction conditions[J].ACS. Catal. 2011,1:116-119.
[14] Liang S, Hammond G B, Xu B. Efficient hydration of alkynes through acid-assisted Bronsted acid catalysis[J].Chem. Commun. 2015,51:903-906.
[15] Tsuchimoto T, Joya T, Shirakawa E, et al. Bronsted acid-catalyzed hydration of alkynes: A convenient route to diverse carbonyl compounds[J].Synlett. 2000,12:1777-1778.
[16] Olivi N, Thomas E, Peyrat J-F, et al. Highly efficient p-toluenesulfonic aicd-catalyzed alcohol addition or hydration of unsymmetrical arylalkynes[J].Synlett. 2004,12:2175-2179.
[17] Liu W, Wang H, Li C-J. Metal-free Markovnikov-type alkyne hydration under mild conditions[J].Org. Lett. 2016,18:2184-2187.
HBF4-Catalyzed Synthesis of Methyl Ketones through Hydration of Terminal Alkynes
YE Dongnai, FU Yejuan, WANG Qinghao, YE Min
(SchoolofChemistryandChemicalEngineering,GannanNormalUniversity,Ganzhou341000,China)
Converting alkynes into carbonyl compounds through hydration is a fundmental transformation in organic synthesis. An efficient HBF4-catalyzed hydration of terminal alkynes to synthesis of methyl ketones has been developed. Trifluoroethanol was used as solvent. We also optimized the reaction conditions, examined the scope of the substrates and proposed the possible mechanisms.
methyl ketone; terminal alkyne; hydration; catalyzed
2016-07-16
10.13698/j.cnki.cn36-1346/c.2016.06.014
国家自然科学基金(21202023);江西省教育厅科技项目(GJJ151005)
叶东鼐(1989-),男,江西安远人,赣南师范大学化学化工学院助教,硕士,研究方向:药物合成及有机合成方法学.
† 通讯作者:叶敏(1982-),女,江西上饶人,赣南师范大学化学化工学院讲师,硕士,研究方向:药物合成、光谱分析.
http://www.cnki.net/kcms/detail/36.1037.C.20161209.1515.030.html
O621
A
1004-8332(2016)06-0062-04
