反应控制相转移催化环戊烯氧化制戊二酸机制研究
2016-12-20张一炯谢志鹏吕志果
周 超,张一炯,谢志鹏,吕志果
(青岛科技大学 化工学院, 山东 青岛 266042)
反应控制相转移催化环戊烯氧化制戊二酸机制研究
周 超,张一炯,谢志鹏,吕志果
(青岛科技大学 化工学院, 山东 青岛 266042)
利用反应控制相转移催化剂[π-C5H5NC16H33]3{PO4[WO4]3}催化环戊烯合成高纯度戊二酸,并对其反应机理和催化剂回收循环机制进行了研究。确定以30%双氧水作为氧化剂时,该催化剂催化环戊烯氧化制戊二酸的较佳反应条件为:CH2O2:Ccpe:C催化剂=4.4:1:0.0037,反应温度90 ℃,反应时间6 h,在该条件下,戊二酸的收率在90%以上。通过GC-MS检测,确定了中间产物和最终产物组成,并以此对反应机理进行了研究。对反应后液体进行重结晶,制得了纯度在99.0%以上的戊二酸产品。利用红外光谱、XRD对新鲜催化剂以及重复利用的催化剂进行了结构表征,研究表明:催化剂在使用过程中活性结构保持不变。催化剂重复使用3次,戊二酸的收率在90%以上。
反应控制相转移催化剂;戊二酸;环戊烯;双氧水
戊二酸是重要的有机化工原料和中间体,在化学、建筑、医药等方面有着广泛的用途。传统的生产戊二酸的方法是从生产己二酸的副产物中回收和利用,随着生产己二酸工艺水平的提高,回收戊二酸的收率和纯度也相应变低[1-4]。另外一种方法是利用硝酸等氧化剂合成戊二酸,这种生产方法对环境的污染大,能耗高,工艺复杂,已经不符合绿色化学的要求[5]。利用传统的均相催化剂例如钨酸,以双氧水作为氧化剂,能够较高收率的合成戊二酸,但是在反应结束后,钨酸等均相催化剂难以从溶液中析出,回收催化剂需要额外的工艺,增加了生产成本[6]。
本研究采用反应控制相转移催化剂,以双氧水为氧源,在较高的收率基础上合成了高纯度戊二酸。探究了在以环戊烯为原料的条件下,合成戊二酸的较佳反应条件,利用 GC-MS检测手段得到了粗产品的组分分布。利用LC检测手段得到了粗产品以及重结晶分离后的样品中戊二酸的纯度。利用红外光谱、XRD对新鲜催化剂及重复利用的催化剂进行了结构表征,得到了催化剂在反应前后结构变化情况,提出了催化剂的合理构成。催化剂进行了多次重复利用,检测催化剂的重复利用效果。对反应控制相转移催化剂催化环戊烯合成戊二酸的反应机理进行了探究。
1 实验部分
1.1 戊二酸合成
称取一定质量的30%双氧水与催化剂粉末于带有冷凝回流管的烧瓶中,48 ℃下水浴加热搅拌,向烧瓶中滴加适量的环戊烯,控制滴加时间为20 min左右,滴加完毕待环戊烯回流结束后,将温度升至90 ℃反应6~8 h。
1.2 戊二酸的分离精制
向反应液中加入适量的甲醛溶液,70 ℃下搅拌1 h,去除反应液中残留的H2O2,回收催化剂固体。将经过处理的反应液利用旋转蒸发仪除去大部分水,0 ℃冷却,得到白色结晶,干燥得到戊二酸样品。
1.3 检测与表征方法
气相质谱检测(GC-MS) GC-MS检测条件为:起始温度80 ℃保持4 min,然后5 ℃/min到150℃,保持4 min,10 ℃/min到300 ℃,保持4 min。进样量0.2 μ L。前ss进样口He,进样口温度320 ℃,分流比 50:1。传输线温度 280 ℃。色谱柱为HP-5MS,325 ℃:30 m×250 μ m×0.25 μ m。
液相检测(LC)室温,Microsorb-MV C18反相柱(4.6 mm×250 mm),紫外检测器,波长定量为247 nm。流动相为0.6 mol/L的苯胍-水溶液+2 mol丁酸,流量为1 ml/L。
X-射线粉末衍射(XRD)采用德国Bruker公司的D8 Advance型X-射线粉末衍射仪进行催化剂样品的物相分析,射线源采用波长为0.15 nm的Cu K α线,采用Goebel镜将发散X光束汇聚为平行光,管电压为40 kV,管电流为150 m A,扫描速率10°/m in。
傅立叶变换红外吸收光谱(FT-IR) FT-IR光谱分析采用德国Bruker公司的Vector-22红外光谱仪,KBr压片,扫描范围为4 000~400 cm-1,扫描次数32次。
1.4 戊二酸收率测定
配置一定浓度的NaOH溶液,称量一定质量的反应液进行滴定,按公式(1)计算戊二酸的收率:
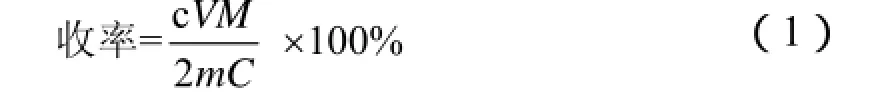
式中:c —NaOH溶液的浓度,mol/L;
V—滴定NaOH溶液的体积,mL;
M—反应液的总质量,g;
m—滴定所用的反应液的质量,g;
C—环戊烯的物质的量,mol。
2 结果与讨论
2.1 反应控制相转移催化机理探究
为了探究反应控制相转移催化环戊烯氧化合成戊二酸的反应机理,进行了以下实验。
在反应过程中,当环戊烯回流结束,升高温度至75 ℃,收集蒸馏出的液体,进行GC-MS检测,得到的组分主要为1,2环戊二醇和戊二醛。
升高温度至 90 ℃后,用异丁醇(微溶于水)萃取,将得到的液体进行气质检测,发现主要的组分为:1,2环戊二醇,戊二醛,戊二酸酐,戊二酸,丁酸丁酯。
48~80 ℃下,将反应产生的气体通入澄清的石灰水,澄清石灰水基本不变浑浊,当温度在90 ℃以上时,将反应产生的气体通入澄清石灰水,澄清石灰水变浑浊[7]。说明温度较低时,C5不会发生脱羧反应,生成CO2和丁酸等C4组分。当温度较高时,戊二酸发生脱羧反应,产生CO2气体和丁酸等副产物。
在48 ℃下环戊烯回流完毕,将温度升至70 ℃反应,反应8~10 h,除水获得粗产品后,对粗产品进行气质检测,发现里面的主要组成成分为:1,2环戊二醇,戊二醛,醛酸C5,戊二酸酐。
利用以上实验结果,可以推测出利用反应控制相转移催化剂,在双氧水的氧化作用下,环戊烯生成戊二酸的反应机理如图1。
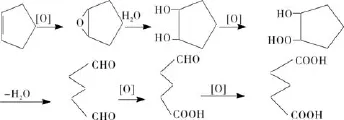
图1 环戊烯氧化生成戊二酸反应机理Fig.1 The reaction mechanism for synthesis of glutaric acid from cyclopentene
在图1中,环戊烯结合了阴离子携带而来的活性氧被环氧化[8-10]。环氧化合物在有水的环境下较难存在,水解生成1,2环戊二醇。因此利用GC-MS难以检测该环氧化合物的其存在。1,2环戊二醇带有两个羟基,在活性氧的作用下,其中一个羟基被氧化生成羧基,氧化后的产物在较高的温度下较容易地脱水开环生成戊二醛,戊二醛的两个醛基被逐步氧化生成戊二酸。如果温度较高(90 ℃以上,当温度升高至90 ℃之后,会有较为明显的反应热效应),少量的戊二酸会发生脱羧反应,生成丁酸。
该催化剂的催化原理为降解聚合反应控制相转移催化[11]。在反应之前,催化剂既不溶于水,也不溶于环戊烯,当具有水溶性的磷钨杂多酸根阴离子{PO4[WO4]3}3-与过氧化氢中的活性氧结合后,降解生成尺寸较小的阴离子,体积较大的阳离子[π-C5H5NC16H33]-可以通过离子对的作用,将体积较小的阴离子萃取到环戊烯中,环戊烯被阴离子中的活性氧氧化。失去活性氧的阴离子再次进入到水相中,与过氧化氢中的活性氧重新结合,再次被萃取到环戊烯中发生氧化作用。当过氧化氢消耗完毕,失去活性氧的阴离子之间聚合生成大尺寸的阴离子,季铵阳离子没有能力再把大体积的阴离子通过离子对的作用萃取到油相中,催化剂在反应溶液中的溶解度大大降低,以沉淀的形式从反应体系中析出。
2.2 氧化反应条件对戊二酸收率的影响
2.2.1 催化剂用量的影响
在原料摩尔比比Ccpe:CH2O2=1;4.4,反应时间t=6 h,反应温度 90 ℃的条件下考察催化剂用量对戊二酸收率的影响(mcpe=14.20 g),结果见图2。从图中可以看出,催化剂的用量从1.0 g增加到1.5 g时,戊二酸的收率从80.0%增加到93.0%,继续增加催化剂的用量,戊二酸的收率基本不发生变化,因此,催化剂的用量为1.5 g较佳。
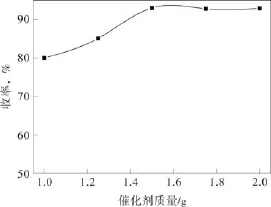
图2 催化剂用量对戊二酸收率的影响Fig.2 Effect of the amount of catalyst on the yield of glutaric acid
2.2.2 反应时间的影响
在原料摩尔比CH2O2:Ccpe:C催化剂=4.4:1:0.0037,反应温度 90 ℃的条件下,考察反应时间对戊二酸收率的影响,结果见图3。
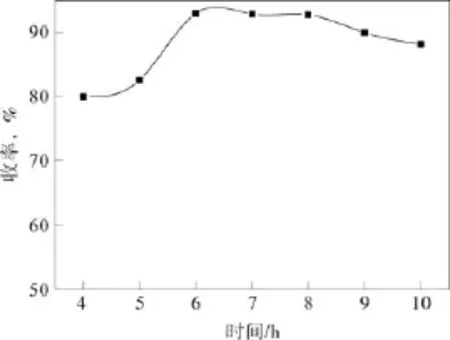
图3 反应时间对戊二酸收率的影响Fig.3 Effect of reaction time on the yield of glutaric acid
从图中可以看出,反应时间从4 h增加到6 h,戊二酸收率从80.0%增加到93.0%,8 h之后,收率不再增加,随着氧化反应时间的延长,少量戊二酸会被深度氧化。因此,较佳的反应时间为6 h。
2.2.3 反应温度的影响
在原料摩尔比CH2O2:Ccpe:C催化剂=4.4:1:0.0037,反应时间6 h的条件下,考察反应温度对戊二酸收率的影响,结果见图4。
从图4中可以看出,从图中可以看出,当温度低于 90 ℃时,戊二酸的收率随着温度的升高而升高,当温度高于 90 ℃时,收率随着温度的升高变化不明显,反而会随着温度的升高略微下降。因此,选择90 ℃为较佳的氧化反应温度。
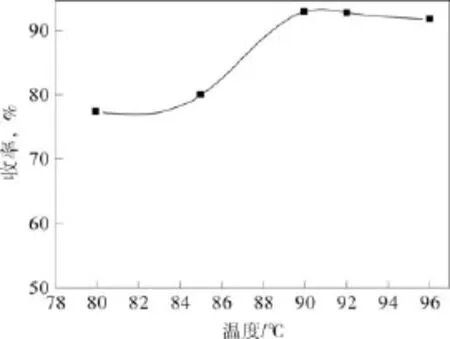
图4 反应温度对戊二酸收率的影响Fig.4 Effect of reaction temperature on the yield of glutaric acid
2.2.4 原料摩尔比的影响
在反应温度90 ℃,反应时间6 h,m催化剂:1.5 g(m环戊烯:14.20 g)的条件下考察原料摩尔比对戊二酸收率的影响,结果见图 5。双氧水在氧化反应过程中,自身会发生部分分解,因此双氧水的用量要高于理论用量。从图中可以看出,当原料摩尔比从4.0增加到4.4过程中。戊二酸收率持续增加,最高为93.0%,继续增加双氧水的用量对戊二酸收率不大。因此,选择CH2O2:Ccpe=4.4:1为较佳的原料摩尔比。
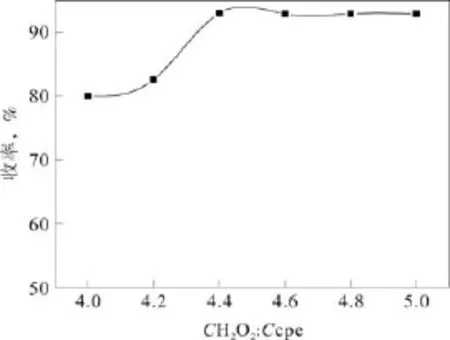
图5 反应物配比对戊二酸收率的影响Fig.5 Effect of matching of reactants on the yield of glutaric acid
通过以上对各影响因素进行分析,得到较佳优化条件为原料摩尔比CH2O2:Ccpe:C催化剂=4.4:1:0.0037,反应温度90 ℃,反应时间6 h。在该条件下,戊二酸的收率为93.0%。
2.3 戊二酸产品的精制与纯度鉴定
2.3.1 产物GC-MS检测
取一定质量的反应液,将水全部蒸干得到浅黄色固体,对该固体进行 GC-MS检测,检测得到除大部分的戊二酸外,还有少量的副产物1,2-环戊二醇、丁酸、戊二酸酐。
2.3.2 产物LC检测
将该浅黄色固体进行LC检测得到谱图如图6。

图6 粗品液相检测Fig.6 The coarse product liquid phase detection
利用LC检测产品中戊二酸的纯度为85.107%。
2.3.3 戊二酸产品的精制
取一定质量的反应液,蒸掉大部分水,重结晶得到白色结晶固体,对该固体进行LC检测,得到谱图如图7。

图7 第一次重结晶液相检测谱图Fig.7 The liquid phase detecting spectra of the first recrystallization
重结晶得到的戊二酸样品进行LC检测,无明显的杂质峰,纯度为99.40%。与纯度为99.0%的戊二酸分析纯液相谱图几乎一致。用熔点仪检测该样品的熔点为:94.7~98.8 ℃,与戊二酸的熔点97.8 ℃相一致。用NaOH溶液滴定纯度为99.29%。用液相检测分析纯戊二酸样品的浓度为99.0%。
2.4 催化剂重复利用考察及其表征
2.4.1 催化剂重复利用考察
取一定质量的回收得到的催化剂按照实验得到的最佳优化条件进行实验,得到的戊二酸收率如表1所示。

表1 回收催化剂对戊二酸收率的影响Table 1 The influence of recycled catalyst on glutaric acid yield
催化剂重复利用3次,戊二酸的收率在90%以上,表明催化剂的重复利用效果较好。
3.4.2 催化剂的红外表征
利用红外光谱对新鲜的催化剂、回用一次的催化剂、回用两次的催化剂进行了表征,得到如下谱图(图8)。

图8 催化剂红外谱图Fig.8 The IR spectra of catalyst
从三种催化剂的红外表征谱图可以看出,经过重复利用的催化剂红外谱图基本相一致,表明该催化剂在经过重复利用之后,结构变化不明显,稳定性较好。新鲜催化剂与经过回收利用的催化剂的红外谱图存在着一定差异,表明该催化剂经过回收利用之后,结构发生了一定的变化。经过分析,在波数3 430处为-NH2或者是游离的-NH,因为催化剂的阳离子含有一个吡啶环,所以会出现此峰。在波数2 921与2 851处为-CH2。不难发现,在波数800处新鲜催化剂与经过回收利用的催化剂之间存在区别,新鲜催化剂在该处振动峰不明显,而经过回收利用的催化剂则在波数 800处有较为明显的振动峰[12],此振动峰属于W-O-W的振动峰,新鲜催化剂中含有磷钨杂多酸根却在此处振动峰不强烈的原因为{PO4[WO4]3}3-离子中WO4处于分离状态,彼此之间没有直接相连,都是与PO43-相连。而催化剂经过利用后,由于磷钨杂多酸根离子充当了携带过氧化氢分子中活性氧的运输工具的作用,在此过程中,磷钨杂多酸根离子结构发生了明显变化,WO4不仅仅能够与PO43-相连,而且W原子之间能够通过O原子共价键相连接。正是由于此种结构的变化,导致催化剂在重复利用时溶于反应体系的速率变慢,但是结构发生变化了的杂多酸跟离子携带活性氧的能力并没有因此减弱,依然保持着较强的催化能力。在波数950处的离子峰为W=O键的伸缩振动峰。波数1 100处的振动峰归属于P-O键的伸缩振动,存在于3种催化剂的杂多酸跟离子中。在波数1 700处的振动峰归属于-C=O的伸缩振动。3.4.3 催化剂的XRD表征
利用XRD对新鲜的催化剂、回用一次催化剂、回用二次催化剂进行了表征,得到如图9谱图。

图9 催化剂XRD检测谱图Fig.9 The XRD spectra of catalysts
从三种催化剂的XRD表征谱图可以发现,新鲜催化剂与经过重复利用的催化剂之间存在着较为明显的结构变化,经过重复利用的催化剂之间谱图几乎相一致,说明催化剂的稳定性较好。新鲜催化剂在 4º~6 º与6º~10 º有较为微弱的衍射峰,而经过重复利用过后的催化剂在4 º ~6 º同样有微弱的衍射峰,在6º~10º有着非常强烈的衍射峰。经过一次重复利用的催化剂在15º~20º,在24º~28º有微弱的衍射峰,而经过两次重复利用的催化剂则不存在。在 20º~24º处,经过重复利用的催化剂都有较为明显的衍射峰。在 6º~10º与 24º~28º代表的是H3PW12O40·6H2O的衍射峰,而在新鲜催化剂中,则不存在此晶体的衍射峰,说明[π-C5H5NC16H33]3{PO4[WO4]3}中的磷钨杂多酸根离子{PO4[WO4]3}3-在经过充当携带活性氧的运输工具后,结构发生变化,该离子部分转化为H3PW12O40·6H2O晶体。而催化剂在经过重复利用后,该种晶体结构依然存在,说明该催化剂具有一定的稳定性。
4 结 论
(1)探究得到了反应控制相转移催化剂[π -C5H5NC16H33]3{PO4[WO4]3}催化双氧水氧化环戊烯合成戊二酸的反应路线、反应机理。
(2)确定反应控制相转移催化剂[π -C5H5NC16H33]3{PO4[WO4]3}催化双氧水氧化环戊烯反应的较佳反应条件为CH2O2:Ccpe:C催化剂=4.4:1:0.0037,反应温度90 ℃,反应时间6 h,在该条件下,戊二酸的收率在90%以上。
(3)利用GC-MS对反应产物进行检测,得到了其组分构成。利用LC等检测方法对重结晶后的样品进行检测,戊二酸的纯度在99.0%以上。
(4)利用XRD、FT-IR表征手段得到了催化剂在反应前后结构的变化情况,发现催化剂在使用过程中活性结构保持不变。催化剂重复利用3次,戊二酸收率均在90%以上。
[1] 沈小洁. 从混合二元酸中分离纯化戊二酸[J]. 广西化工, 1998, 27(4): 34-35.
[2] Pehlvanoglu N, Uslu H, Kirbaslar S I. Experimental and modleling Studies on the Extraction of Glutaric Acid by Trioctylamine[J]. J. Chem. Eng. Data, 2009, 54(12): 3202-3207.
[3] Qin W, Zhang Y, Li Z, et al. Extraction Equilibria of Glocolic and Glyoxylic Acids with Trialkylphosphine Oxide and Trioctylamine as Extractant[J]. J. Chem. Eng. Data, 2003, 48(2): 430-434.
[4] Pehlvanoglu N, Uslu H, Kirbaslar S I. Extractive Separation of Glutaric Acid by Aliquat 336 in Different Solvents[J]. J. Chem. Eng. Data, 2010, 55(9): 2970-2973.
[5] 华文. γ-丁内酯的生产技术和市场分析[J]. 精细化工原料及中间体, 2006 (5): 39-42.
[6] 陈慧. 戊二酸绿色合成工艺及新型催化剂研究[D]. 上海: 复旦大学, 2007.
[7] 龚艳, 林鹿, 孙勇, 庞春生. 脱羧反应途径及其机制的研究进展[J]. 化学与生物工程, 2008, 25(4): 1-6.
[8] 张术栋, 徐成华, 等. 烯烃环氧化及其催化剂的研究进展[J]. 合成化学, 2003(4): 294-299.
[9] 李丽, 彭军, 张龙. 杂多化合物催化过氧化氢氧化烯烃环氧化反应研究进展[J]. 精细化工中间体, 2006, 36(5): 24-27.
[10] 陈杨英, 韩秀文. 过氧化氢为氧源催化烯烃环氧化研究[J]. 化学进展, 2006, 18(4): 399-402.
[11] 赵地顺. 相转移催化原理及应用[M]. 北京: 化学工业出版社, 2007.
[12] Radkove., Beer R. H. High yield synthesis of mixed-metal keggin polyoxoanions in non-aqueous solvents: Preparation of (n-Bu4N)4 [PMW11O40](M=V,Ng,Ta)[J]. Polyhedron, 1995, 14(15-16): 2139-2143.
Study on Synthesis of Glutaric Acid From Cyclopentene Over Reaction-controlled Phase Transfer Catalyst
ZHOU Chao,ZHANG Yi-jiong,XIE Zhi-peng,LV Zhi-guo
(School of Chemical Engineering,Qingdao University of Science and Technology, Shandong Qingdao 266042,China)
Glutaric acid with high purity was synthesized from cyclopentene by using reaction-controlled phase transfer catalyst [π-C5H5NC16H33]3{PO4[WO4]3}, the reaction and catalyst recycling mechanisms were also studied. The suitable reaction condition was determined as follow: CH2O2:Ccpe:Ccatalyst=4.4:1:0.0037, the reaction temperature 90 ℃, reaction time 6 h, using 30% hydrogen peroxide as oxidant. Under this condition, the yield of glutraric acid can be over 90%. The compositions of intermediates and final products were determined by GC-MS testing, the reaction mechanism was proposed. The products with more than 99.0% purity could be obtained by recrystallization of the liquid after the reaction. The activity structure of the catalyst kept invariability in the process of using it. The yield of glutaric acid was over 90% in reusing of the catalyst.
reaction-controlled phase transfer catalyst; glutaric acid; cyclopentene; hydrogen peroxide
TQ 201
A
1671-0460(2016)11-2547-05
2016-05-25
周超(1990-),男,山东省莱芜市人,硕士研究生,就读于青岛科技大学化学工程与技术专业,研究方向:精细化工。E-m ail:1044549484@qq.com。
吕志果(1967-),男,教授,博士学位,研究方向:精细化工。E-m ail:guoguolv@sina.com。
