基于 454高通量测序的黄土高原不同乔木林土壤细菌群落特征
2016-12-20曾全超黄懿梅西北农林科技大学资源环境学院陕西杨凌712100
刘 洋,曾全超,黄懿梅(西北农林科技大学资源环境学院,陕西 杨凌 712100)
基于 454高通量测序的黄土高原不同乔木林土壤细菌群落特征
刘 洋,曾全超,黄懿梅*(西北农林科技大学资源环境学院,陕西 杨凌 712100)
选取黄土高原不同乔木林(辽东栎, LDL;侧柏, CB;刺槐, CH;油松, YS)表层土壤为研究对象,利用第二代高通量测序技术罗氏454平台对其进行16S rRNA基因V1~V3可变区测序,通过分析其Alpha多样性、物种组成和丰度、群落结构,结合土壤的理化性质研究其对细菌群落结构的影响.结果表明:所有土壤样品共检测到36个门,84个纲,187个目.不同乔木林土壤中优势菌来自变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、绿弯菌(Chloroflexi)和浮霉菌门(Planctomycetes),主要的优势菌纲为放线杆菌纲(Actinobacteria)、α-变形菌纲(Alphaproteobacteria)、酸杆菌(Acidobacteria)、β-变形菌纲(Betaproteobacteria)、浮霉菌纲(Planctomycetacia).不同乔木林土壤中绿弯菌门(Chloroflexi)与pH值呈极显著的正相关,总磷和蓝细菌(Cyanobacteria)呈极显著的正相关,微生物生物量碳与芽单胞菌门(Gemmatimonadetes)呈极显著的负相关,土壤总磷含量可能为CB样地区别于其他土样群落组成的主要因素. LDL样地细菌群落受环境影响较小.
454高通量测序;子午岭;不同乔木林;土壤细菌;环境因子
土壤是一个非常复杂的环境体系,而土壤微生物是联系土壤与植物的纽带,同时土壤微生物还参与土壤中碳、氮循环,对于全球气候变化有着不忽略的影响[1-2].由于土壤微生物物种丰富、难以培养、功能多样,土壤中可培养微生物仅占到土壤整个微生物群落的 1%~10%,通过传统的纯培养方法不能准确、全面地反映土壤微生物群落结构与分布状况[4].21世纪以来,新一代高通量测序技术的快速发展,可直接测序16S rRNA基
因的PCR产物,每次测序可获得数以百万甚至亿万条基因序列,由于其通量高、反应灵敏、序列数多,是研究土壤微生物群落结构、功能特性的重要技术手段[5].目前第二代测序技术已经成为研究微生物的重要技术手段.相比传统的测序方法,高通量测序技术能够更加准确全面地反映土壤微生物群落结构特征[6],已经被众多学者应用于土壤生态系统的研究[7-11].
黄土高原作为典型退化生态系统的代表,同时也是中国乃至世界重大的侵蚀区.1999年来,我国在黄土高原开展了大规模的植树造林、退耕还林还草生态工程,并取得了显著的成效,植被覆盖度与土壤质量都得到了不同程度的改善[12].而土壤微生物在这过程起着非常重要的作用.目前,研究黄土高原不同植被类型土壤微生物主要采用的是传统的纯培养法[13]、磷脂脂肪酸法[14-15]、PCR-RFLP 等方法[16],由于以上传统方法的局限性,不能准确直观地解析土壤微生物的变化规律.因此,本研究选取黄土高原子午岭林区四种典型乔木林的土壤为研究对象,采用罗氏454焦磷酸高通量测序方法,研究其土壤微生物多样性,探讨植被恢复对土壤微生物多样性的影响,以期为黄土高原植被恢复和生态系统构建提供参考依据.
1 材料与方法
1.1 研究区概况
子午岭地跨陕西、甘肃2省,处于黄土丘陵区的腹地,子午岭林区是黄土丘陵区目前保存较好的天然植被区,是黄土丘陵区中部地带重要的次生原始森林.子午岭地理坐标为 107°30′~109°40′E,33°50′~36°50′N.地势南高北低,自西向东北倾斜,海拔为 1300~1700m,该区域处于森林草原和半干旱草原的过渡区,气候温和湿润,其北小半部属陇中北部温带半干旱气候,南大半部属陇中南部温带半湿润气候,年平均气温为 7.4~8.5℃,极端最低温度为-27.7℃,极端最高气温为36.7℃,≥10℃的活动积温 2671.0℃,无霜期110~150d,平均降水量 587.6mm,干燥度 0.97,平均相对湿度 63%~68%,地带性土壤以石灰性灰褐土为主.
1.2 样品采集
样品于2013年8月采集自中科院水土保持研究所子午岭土壤侵蚀与生态环境观测站附近,选择4种年龄相近(15a左右)的典型乔木林,分别为辽东栎、侧柏、油松、刺槐,样地基本信息见表 1.每种乔木林下,都选择地形较为相似的3块样地作为野外重复,每块样地相距至少 2.5km,在各个样地内设置3个20m×20m的样方,在每一个样方内由下向上按S形布点法选5个采样点,在各点利用土钻采集0~5cm的土样,去除根系、石块、混匀后分成2份,1份立即放入-80℃的冰箱里保存用于DNA的提取,1份存储于4℃冰箱中,用于测定土壤的理化性质.

表1 不同乔木林样地描述Table 1 The description of sample sites in different arbors forests
1.3 土壤基本特性分析
土壤有机质、全氮、总磷、容重、含水率均按照《土壤农化分析》的标准方法进行分析测定[17].土壤微生物生物量碳氮采用氯仿熏蒸0.5mol/LK2SO4浸提法[18-19].其中浸提液中的溶解性碳(DOC)采用总有机碳分析仪(Phoenix 8000,美国) 测定,由熏蒸与未熏蒸土样的 DOC差值计算得到微生物生物量碳 (MBC);浸提液中溶解性氮 (DON)采用碱性过硫酸钾氧化法测定[20](UV 2800A),熏蒸与未熏蒸土样的DON的
差值得到微生物生物量氮(MBN).微生物生物量碳氮的转换系数均为0.45[21].
1.4 土壤微生物DNA的提取
每种土壤样品称取0.5g用于DNA提取,使用OMEGA 公司 E.Z.N.A Soil DNA试剂盒抽提基因组DNA.完成DNA的提取后,利用1%琼脂糖凝胶电泳检测抽提的基因组DNA.
1.5 PCR扩增与焦磷酸 454测序
PCR 采用 TransGen AP221-02:TransStart Fastpfu DNA Polymerase;PCR 仪 :ABI GeneAmp® 9700型;全部样品按照正式实验条件进行,每个样品3个重复,将同一样品的PCR产物混合后用 2%琼脂糖凝胶电泳检测,使用AxyPrepDNA凝胶回收试剂盒(AXYGEN公司)切胶回收PCR产物,Tris_HCl洗脱;2%琼脂糖电泳检测.参照电泳初步定量结果,将 PCR产物用QuantiFluor™-ST蓝色荧光定量系统(Promega公司)进行检测定量,之后按照每个样品的测序量要求,进行相应比例的混合.
按指定测序区域,合成带有“5’ 454A、B接头-特异引物 3’”的融合引物.引物为 27F(5’-AGAGTTTGATCCTGGCTCAG-3’)和 533R(5’-TTACCGCGGCTGCTGGCAC-3’)[6]用Roche GS FLX+ Sequencing Method Manual_XLR70kit(上海美吉生物有限公司)进行上机测序.
1.5 数据处理
1.5.1 基本数据处理 所有数据经过 Excel 2013处理,利用SPSS 20.0进行单因素方差分析析(One-Way ANOVA)和最小显著差异法(LSD)进行分析比较.
1.5.2 测序数据的分析与处理 (1)序列的优化及去杂
根据 barcode 序列区分各个样品的测序数据,提取的数据以 sff(Standard flowgram format)格式保存,sff文件属二进制文件,可以提取出fasta序列文件和qual质量文件.高通量测序中通常会出现一些点突变和高分子均聚物等测序错误,随着测序长度的增加序列末端的质量会降低,为了得到更高质量及更精准的生物信息分析结果,则应对有效序列进行去杂和修剪得到优化数据.使用软件Mothur(vsesion 1.17http://qiime. org/)对数据去杂[22].
(2)OTU聚类
OTU(Operational Taxonomic Units)是在系统发生学或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,属,种、分组等)设置的同一标志.要了解一个样本测序结果中微生物的种属信息,就需要对序列进行归类操作.通过归类操作,将序列按照彼此的相似性分归为许多小组,一个小组就是一个 OTU.可根据不同的相似度水平,对所有序列进行 OTU 划分,通常在97%的相似水平下的OTU进行生物信息统计分析.软件平台:Usearch(vsesion 7.1http://drive5. com/uparse/).
(3)分类学分析
为了得到每个OTU 对应的物种分类信息,采用 RDP classifier 贝叶斯算法对 97%相似水平的OTU 代表序列进行分类学分析,并分别在各个分类水平(门,纲,目,科,属,种)统计各样本的群落组成.比对数据库如下:16S细菌和古菌核糖体数据库(Silva(Release119http://www.arbsilva. de); RDP(Release 11.1 http://rdp.cme.msu. edu/) Greengene (Release13.5http://greengenes. secondgenome. com/).
(4)多样性指数分析
群落生态学中研究微生物多样性,通过单样本的多样性分析可以反映微生物群落的丰度和多样性,包括一系列统计学分析指数估计环境群落的物种丰度和多样性.
计算菌群丰度的指数有:①Chao:是用 chao1算法估计群落中含OTU数目的指数,chao1在生态学中常用来估计物种总数,由Chao (1984)最早提出.②Ace:用来估计群落中含有OTU数目的指数,由 Chao提出,是生态学中估计物种总数的常用指数之一,与Chao的算法不同.
计算菌群多样性(Community diversity)的指数有:①Simpson:用来估算样品中微生物的多样性指数之一,由Edward Hugh Simpson (1949)提出,在生态学中常用来定量的描述一个区域的生物多样性.Simpson指数值越大,说明群落多样性
越低.②Shannon:用来估算样品中微生物的多样性指数之一.它与Simpson多样性指数均为常用的反映alpha多样性的指数.Shannon值越大,说明群落多样性越高.
测序深度指数有:Coverage:是指各样品文库的覆盖率,其数值越高,则样本中序列没有被测出的概率越低.该指数实际反映了本次测序结果是否代表样本的真实情况.
对OTU列表中获得的分类信息与丰度进行整理,在门和纲分类水平下对各样品进行物种丰度统计及 RDA分析,可得到样品中群落组成结构、相似性以及群落结构与环境因子的关系.其中 RDA(基于线性模型)分析图是一种基于对应发展的排序方法,将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析,可以反映群落组成与环境因子之间的关系.
2 结果与分析
2.1 土壤理化性质
不同乔木林下土壤的基本理化性质见表 2.子午岭区 4种乔木林土壤为弱碱性,pH值在 7.84~ 8.17之间变动,不同植被之间差异不显著(P>0.05).土壤容重为0.69~1.10g/cm3,LDL>CB> YS>CH.土壤含水率LDL最高,其他3种乔木林变化不大.土壤有机碳不同植被之间差异显著,LDL>CB>YS>CH.土壤全氮表现出与土壤有机碳相同的变化趋势,LDL显著高于其他植被群落(P<0.05).

表2 不同乔木林下土壤的基本特性Table 2 Soil biochemical properties under different arbors forests
如表 3所示,不同乔木林土壤 MBC为 350.83~693.15mg/kg,且含量大小顺序为 LDL> CB>YS>CH,其中 LDL和 CB显著高于 YS、CH(P<0.05).土壤MBN在52.21~93.61mg/kg之间变化,大小顺序为:LDL>CB>YS>CH,LDL和CB显著大于YS和CH.4种乔木林土壤微生物量碳氮比值(MBC/MBN)大体在6~8之间变化,比值大小顺序为 YS(7.76)>LDL(7.39)>CH(6.73)>CB(6.72),不同乔木林土壤微生物量氮占全氮的比例在 3.21%~5.03%之间变化,微生物量碳占总有机碳比例为 2.02%~3.29%.土壤碳氮比(SOC/ TN)为8.46~12.42,CH最低,LDL最高.

表3 不同乔木林下土壤的微生物生物量碳氮以及比值Table 3 Soil microbial biomass under different arbors forests
2.2 Alpha 多样性分析结果
如表 4所示,4种样品分别获得有效序列数为10405、8500、8601、9306,样品的平均覆盖率为90%,且稀释曲线趋于平台期,表明该测序效果
理想.在 97% 分类水平下,黄土高原不同乔木林下微生物 Chao、Ace、Simpson指数、Shannon指数、OTU数量有所差异.Chao指数大小顺序为:YS> CH > CB >LDL;Ace指数为YS> CH > CB>LDL,Shannon指数大小顺序为:YS>CB>CH>LDL;Simpson指数大小顺序为:LDL>CH>CB>YS.OUT数目为 1969~2435,YS>CB>CH>LDL. YS乔木林下土壤微生物的多样性最高,LDL土壤微生物多样性最低.

表4 序列统计及多样性指数Table 4 Sequence statistics and diversity index
2.3 群落结构组成分析
如图1A 所示,通过454高通量测序发现4种黄土高原不同乔木林下的土壤中检测到的主要微生物有:放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)、芽单胞菌门(Gemmatimonadetes)、装甲菌门(Armatimonadetes)、蓝菌门(Cyanobacteria)等.变形 菌 门 (Proteobacteria)、 放 线 菌 门(Actinobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)是土壤中占主导地位的微生物类群,约占到了所有微生物总数的 80%~85%以上.不同乔木林下放线菌门(Actinobacteria)的相对丰度变化范围为12.66%~19.15%,YS>CB>LDL>CH;变 形菌门(Proteobacteria)的 变 化 范 围 为 29.40%~37.90%,CB>CH>LDL>YS;绿弯菌门(Chloroflexi)的变化范围为9.25%~14.67%,CH>YS>LDL>CB;酸杆菌门(Acidobacteria)的变化范围为 8.51%~20.21%,LDL>YS>CB>CH; 浮 霉 菌 门(Planctomycetes)的变化范围为 7.99%~10.02%, YS>CH>LDL>CB; 芽 单 胞 菌 门(Gemmatimonadetes)的变化范围为3.05%~6.27%, CH>YS>CB>LDL.
如图 1B所示,在纲分类水平上,不同乔木林下土壤主要的优势菌分别为放线杆菌纲(Actinobacteria)、α-变形菌纲(Alphaproteobacteria)、酸 杆 菌 (Acidobacteria)、 β-变 形 菌 纲(Betaproteobacteria)、浮霉菌纲(Planctomycetacia) (所占的比例分别为 12.66%~ 19.15%, 15.00%~24.11%, 8.51%~20.21, 4.61%~11.68%, 4.38%~5.36%.α-变形菌纲(Alphaproteobacteria)在 CB、LDL样地分布较多,β-变形菌纲(Betaproteobacteria)在 YS、CH 样地分布较多.浮霉菌纲(Planctomycetacia)、厌氧绳菌纲(Anaerolineaceae)、 Phycisphaerae、δ-变形菌纲(Deltaproteobacteria)在不同乔木林下差异较小.
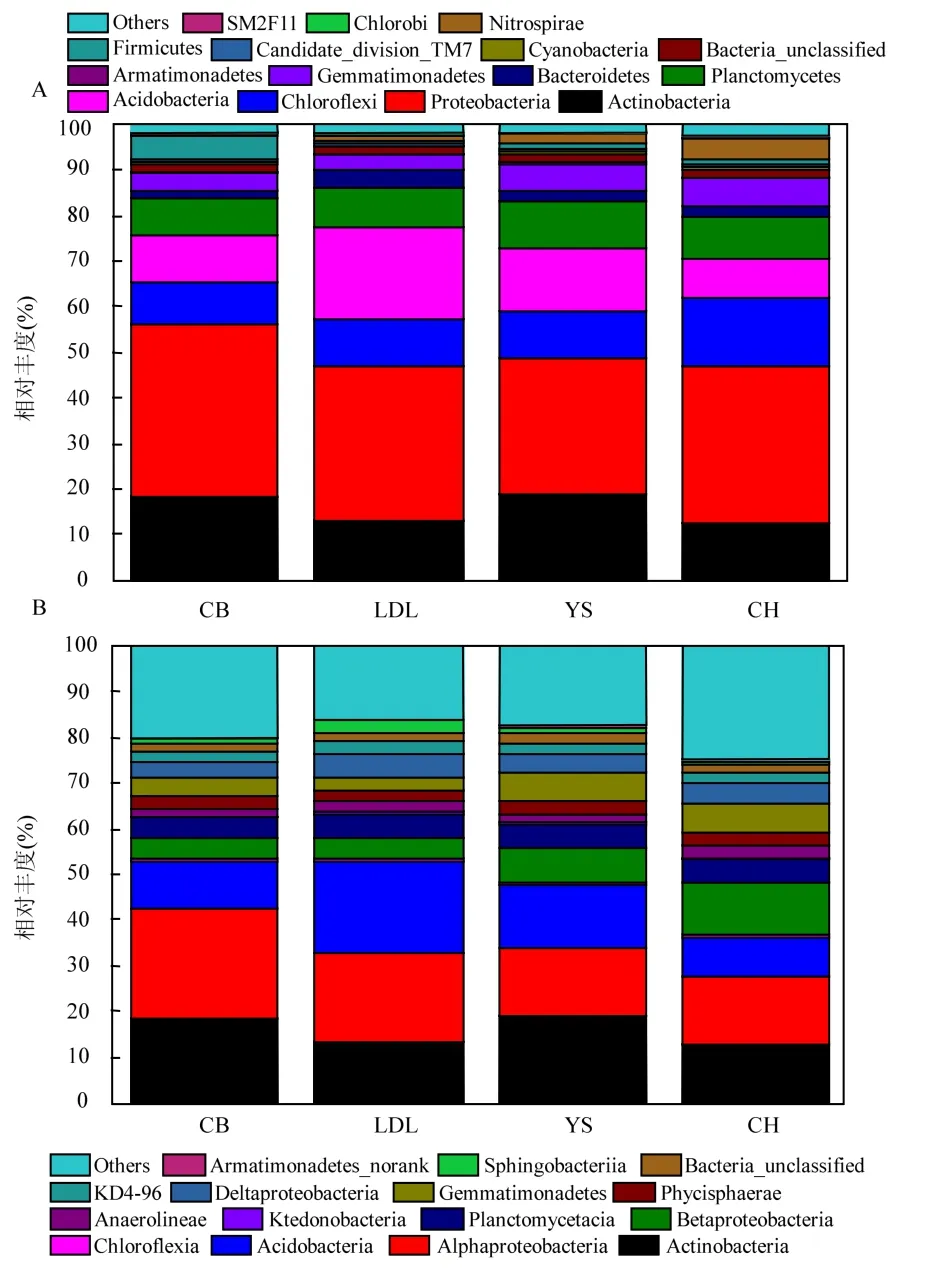
图1 门和纲分类水平下的微生物群落组成Fig.1 The relative richness of soil bacterial community at phylum (A) and class (B) levels
2.4 环境因素对微生物群落的影响
土壤细菌在门分类水平上的相对丰度与土壤理化性质的关系如图 2 所示.从图 2 中可看出,CB 和LDL 与YS、CH 土样的微生物群落结构差异较大,土样分别位于第一、二、三象限.YS与 CH土壤的微生物物种分布差异较小,丰度较高的是绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)、酸杆菌门(Acidobacteria)、芽单胞菌门(Gemmatimonadetes)在 LDL样地分布较多.pH值与绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)群落正相关,TP与变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、蓝菌门(Cyanobacteria)群落呈负相关.
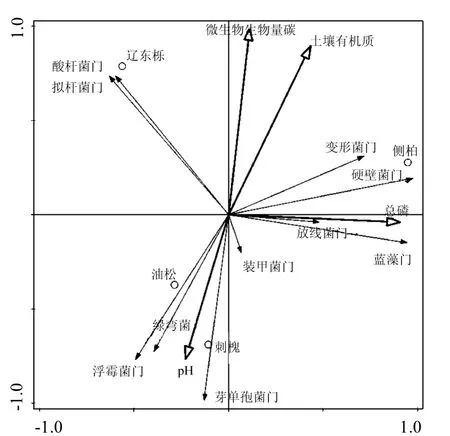
图2 基于土样和环境因子的冗余分析Fig.2 Redundancy analysis (RDA) based on soil bacterial community at phylum level and environmental factors of the soil samples
3 讨论
已有研究表明,黄土高原经过近20年的植被恢复,土壤的物理、化学、生物性质已经发生了显著的改善.植被恢复通过枯枝落叶以及根系能够有效地提高土壤的碳、氮、磷等养分含量[23],从而影响土壤微生物区系的形成[24].此外,植被恢复能够提高土壤的团聚体的的稳定性[25],在水土保持与生态服务功能方面起着重要的作用.本研究中发现不同乔木林下MBC/MBN在6~8 之间变化,略低于赵彤等[26]研究的人工乔木林下的微生物量碳氮比(9~11),与李香珍等[27]研究的5~9接近.有研究表明,土壤中MBC/MBN可以很好地反映土壤中微生物的种类和区系[28],其中细菌的碳氮比为 5:1,真菌的碳氮比 10:1,放线菌的碳氮比为6:1[29].但是MBC/MBN只能粗略的判定土壤中细菌、放线菌、真菌的分布情况,不能准确评价土壤细菌在各个分类水平上的分布以及相对丰度.造成不同乔木林下土壤MBC/MBN差异的主要原因是植被的凋落物和根系物分解,导致不同的微生物区系形成[30],植被还可以通过对土壤水分、土壤养分、pH值的影响,改变土壤中的微生物组成[31].由于不同植被类型下土壤养分的不同,可以形成不同的微生物群落结构[32],因此植被类型对土壤微生物量产生较大的影响.
在微区域内,土壤微生物的生长主要受土壤的物理化学性质的影响.微生物通过分解经过多年的累积枯枝落叶来影响土壤养分循环及其自身的多样性[11].有研究表明,放线菌门(Actinobacteria)是降解木质素[33]与纤维素[34]的主要功能菌门.4种乔木林土壤放线菌门(Actinobacteria)所占比例范围在 12.66%~19.15%之间,与杨树人工林土壤较为一致[10].研究表明植物群落类型是影响土壤放线菌多样性的重要因素[35-36].在变形菌门(Proteobacteria)中,α-变形菌纲(Alphaproteobacteria)是最主要的纲,其次是 β-变形菌纲(Betaproteobacteria),所占比例分别为 14.65%~24.11%,4.61%~11.68%,这与韩亚飞等[10]人对杨树人工林等的研究一致,而Roesch等[8]和Zhang等[9]研究发现 β-变形菌纲(Betaproteobacteria)的丰富度大于α-变形菌纲(Alphaproteobacteria).土壤pH是影响微生物群落分布的主要影响因素[37-39].在盐碱土中,α-变形菌纲(Alphaproteobacteria)、β-变形菌纲(Betaproteobacteria)、γ-变形菌纲(Gammaproteobacteria)和δ-变形菌纲(Deltaproteobacteria)是最重要的微生物类群[35].本研究中 4种乔木林下,变形菌门(Proteobacteria)是最主要的优势菌门,所占比例为 24.90%~ 37.90%,而该地区土壤呈弱碱性,也验证了变形菌门(Proteobacteria)为碱性土壤中的主要优势群落.Liu等[6]发现影响东北黑土土壤微生物地理分布的关键因子是土壤 pH值,这与本
研究一致.土壤碳氮磷含量是土壤微生物生长的主要来源,不同的肥力状况也会影响土壤微生物种群数量及分布.LDL样地的土壤有机质含量、总氮、总磷、微生物量碳氮最高,酸杆菌门(Acidobacteria)、拟杆菌门(Bacteroidetes)的相对丰富度也是最高的,可能养分差异所导致的.有研究发现土壤有机碳含量是驱动土壤微生物生物地理学分布的主要因素[6].不同乔木林下土壤的有机碳差异显著,土壤有机碳可能也是影响不同乔木林土壤细菌分布的主要因子.综合全国其他地区应用高通量测序方法的研究发现,东北黑土与黄土高原乔木林土壤的主要优势菌门均为酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes).但是不同的生态系统中,主要的优势菌群的相对丰度存在差异.在东北黑土中,优势细菌为酸杆菌门(Acidobacteria) (24.11%)与变形菌门(Proteobacteria)(19.25%),其中土壤有机碳与土壤pH是影响东北黑土细菌生物地理分布的主要环境因子[6].
目前,高通量测序方法已经成为研究不同地区土壤微生物的生物地理学分布的主要方法[6-7,38].对于纬度是否是影响土壤微生物分布的主要原因,不同的研究者得出不同的结论[21,40-41].黄土高原是中国最大的黄土侵蚀区,其土壤微生物的生物地理学分布,尤其是不同植被类型下土壤碳氮循环的功能微生物分布研究较少,这也是我们后面的研究的重点.
4 结论
4.1 利用454高通量测序技术分析不同林型土壤样品,平均覆盖率为90%,测序结果能够全面的反映样品组成及结构.多样性指数表明4种林型土壤细菌的物种丰富度顺序为 YS>CB>CH>LDL. YS土壤的细菌多样性最高, LDL细菌多样性最低.
4.2 不同乔木林土壤中优势菌门为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、绿弯菌(Chloroflexi)、浮霉菌门(Planctomycetes),在纲分类水平上主要的优势菌为放线杆菌纲(Actinobacteria)、α-变形菌纲(Alphaproteobacteria)、酸杆菌(Acidobacteria)、β-变形菌纲(Betaproteobacteria)、浮霉菌纲(Planctomycetacia),这些门占总体的80%以上.
4.3 不同乔木林土壤细菌的相对丰度与土壤理化性质程序显著的相关性,其中绿弯菌门(Chloroflexi)与pH呈极显著的正相关,TP和蓝藻细菌呈极显著的正相关,MBC与芽单胞菌门(Gemmatimonadetes)呈极显著的负相关, TP含量可能为 CB样地区别于其他土样群落组成的主要因素. LDL样地细菌群落受环境影响较小.
[1] Harris J. Measurements of the soil microbial community for estimating the success of restoration [J]. European Journal of Soil Science, 2003,54(4):801-808.
[2] 杨 宁,邹冬生,杨满元,等.衡阳紫色土丘陵坡地恢复过程中土壤微生物生物量与土壤养分演变 [J]. 林业科学, 2014,50(12):144-150.
[3] 于贵瑞,高 扬,王秋凤,等.陆地生态系统碳氮水循环的关键耦合过程及其生物调控机制探讨 [J]. 中国生态农业学报, 2013, 21(1):1-13.
[4] 张洪霞,谭周进,张祺玲,等.土壤微生物多样性研究的dggeπtgge技术进展 [J]. 核农学报, 2009,23(4):721-727.
[5] 夏围围,贾仲君.高通量测序和DGGE分析土壤微生物群落的技术评价 [J]. 微生物学报, 2014,54(12):1489-1499.
[6] Liu J J, Sui Y Y, Yu Z H, et al. High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China [J]. Soil Biology and Biochemistry, 2014,70:113-122.
[7] Liu W H, Zhu J J, Jia Q Q, et al. Carbon sequestration effects of shrublands in three-north shelterbelt forest region, China [J]. Chinese Geographical Science, 2014,24(4):444-453.
[8] Roesch L F, Fulthorpe R R, Riva A, et al. Pyrosequencing enumerates and contrasts soil microbial diversity [J]. The ISME Journal, 2007,1(4):283-290.
[9] Zhang T, Shao M-F, Ye L. 454pyrosequencing reveals bacterial diversity of activated sludge from 14sewage treatment plants [J]. The ISME Journal, 2012,6(6):1137-1147.
[10] 韩亚飞,伊文慧,王文波,等.基于高通量测序技术的连作杨树人工林土壤细菌多样性研究 [J]. 山东大学学报(理学版), 2014, 49(5):1-6.
[11] 井赵斌,程积民,张宝泉,等.基于 454焦磷酸测序法的典型草原土壤真核生物多样性 [J]. 草业科学, 2013,30(11):1690-1697.
[12] Deng L, Liu G, Shangguan Z. Land use conversion and changing soil carbon stocks in China's ‘grain-for-green’ program: A synthesis [J]. Global Change Biology, 2014,20(11):3544-3556.
[13] 邢肖毅,黄懿梅,安韶山,等.黄土丘陵区不同植被土壤氮素转化微生物生理群特征及差异 [J]. 生态学报, 2013, 33(18):5608-5614.
[14] 胡婵娟,郭 雷,刘国华.黄土丘陵沟壑区不同植被恢复格局下土壤微生物群落结构 [J]. 生态学报, 2014,34(11):2986-2995.
[15] 刘海燕,魏天兴,王 仙.黄土丘陵区人工林土壤微生物PLFA标记多样性分析 [J]. 北京林业大学学报, 2016,38(1):28-35.
[16] 刘桂婷,程 林,王保莉,等.长期不同施肥对黄土旱塬黑垆土氨氧化细菌多样性的影响 [J]. 中国农业科学, 2010,43(13):2706-2714.
[17] 鲍士旦.土壤农化分析 [M]. 北京:中国农业出版社, 2000.
[18] Sparling G P, West A W. Modifications to the fumigation- extraction technique to permit simultaneous extraction and estimation of soil microbial-c and microbial-n [J]. Communications in Soil Science and Plant Analysis, 1988,19(3):327-344.
[19] Vance E D, Brookes P C, Jenkinson D S. An extraction method for measuring soil microbial biomass-c [J]. Soil Biology and Biochemistry, 1987,19(6):703-707.
[20] Cabrera M L, Beare M H. Alkaline persulfate oxidation for determining total nitrogen in microbial biomass extracts [J]. Soil Science Society of America Journal, 1993,57(4):1007-1012.
[21] Jenkinson D S, Brookes P C, Powlson D S. Measuring soil microbial biomass [J]. Soil Biology and Biochemistry, 2004, 36(1):5-7.
[22] Schloss P D, Westcott S L, Ryabin T, et al. Introducing mothur:Open-source, platform-independent, community-supported software for describing and comparing microbial communities [J]. Applied and Environmental Microbiology, 2009,75(23):7537-7541.
[23] 曾全超,李 鑫,董扬红,等.陕北黄土高原土壤性质及其生态化学计量的纬度变化特征 [J]. 自然资源学报, 2015,30(5):870-879.
[24] Huang Y, Michel K, An S,et al. Changes in microbial-community structure with depth and time in a chronosequence of restored grassland soils on the loess plateau in northwest China [J]. Journal of Plant Nutrition and Soil Science, 2011,174(5):765-774.
[25] An S, Mentler A, Mayer H, et al. Soil aggregation,aggregate stability,organic carbon and nitrogen in different soil aggregate fractions under forest and shrub vegetation on the loess plateau, china [J]. Catena, 2010,81(3):226-233.
[26] 赵 彤,闫 浩,蒋跃利,等.黄土丘陵区植被类型对土壤微生物量碳氮磷的影响 [J]. 生态学报, 2013,33(18):5615-5622.
[27] 李香真,曲秋皓.蒙古高原草原土壤微生物量碳氮特征 [J]. 土壤学报, 2002,39(1):97-104.
[28] 黄昌勇.土壤学 [M]. 北京:中国农业出版社, 2000.
[29] Cleveland C C, Liptzin D. C: N: P stoichiometry in soil: Is there a“redfield ratio” for the microbial biomass [J]. Biogeochemistry, 2007,85(3):235-252.
[30] 姜培坤,周国模.侵蚀型红壤植被恢复后土壤微生物量碳,氮的演变 [J]. 水土保持学报, 2003,17(1):112-114.
[31] 汪杏芬,李世仪.CO2培增对植物生长和土壤微生物生物量碳,氮的影响 [J]. 植物学报:英文版, 1998,40(12):1169-1172.
[32] 任建宏,燕辉,朱铭强,等.秦岭北坡 4种植被类型的土壤养分状况和微生物特征比较研究 [J]. 水土保持研究, 2010,17(4):228-232.
[33] Lynd L R, Weimer P J, Van Zyl W H, et al. Microbial cellulose utilization: Fundamentals and biotechnology [J]. Microbiology and Molecular Biology Reviews, 2002,66(3):506-577.
[34] Pankratov T A, Ivanova A O, Dedysh S N, et al. Bacterial populations and environmental factors controlling cellulose degradation in an acidic sphagnum peat [J]. Environmental Microbiology, 2011,13(7):1800-1814.
[35] 刘绍雄,王明月,王 娟,等.基于PCR-DGGE技术的剑湖湿地湖滨带土壤微生物群落结构多样性分析 [J]. 农业环境科学学报, 2013,32(7):1405-1412.
[36] 张晓红,胡文革,莫 超,等.艾比湖湿地根际放线菌多样性及其环境响应 [J]. 环境科学与技术, 2015,38(12):22-30.
[37] 李 新,焦 燕,代 钢,等.内蒙古河套灌区不同盐碱程度的土壤细菌群落多样性 [J]. 中国环境科学, 2016,36(1):249-260.
[38] Fierer N, Jackson R B. The diversity and biogeography of soil bacterial communities [J]. Proceedings of the National Academy of Sciences of the United States of America, 2006,103(3):626-63.
[39] 袁红朝,吴 昊,葛体达,等.长期施肥对稻田土壤细菌,古菌多样性和群落结构的影响 [J]. 应用生态学报, 2015,26(6):1807-1813.
[40] Chan O C, Yang X, Fu Y, et al. 16s rRNA gene analyses of bacterial community structures in the soils of evergreen broad-leaved forests in south-west china [J]. Fems Microbiology Ecology, 2006,58(2):247-259.
[41] Terhonen E, Marco T, Sun H, et al. The effect of latitude, season and needle-age on the mycota of scots pine (pinus sylvestris) in Finland [J]. Silva Fennica, 2011,45(3):301-317.
致谢:感谢董扬红、李鑫、李娅芸同学帮忙采样,感谢上海美吉生物有限公司技术人员的数据分析与处理.
Soil microbial communities by 454prosequencing under different arbor forests on the Loess Plateau.
LIU Yang, ZENG Quan-chao, HUANG Yi-mei*(College of Natural Resources and Environment, Northwest A&F University, Yangling 712100, China). China Environmental Science, 2016,36(11):3487~3494
To determine the diversity of soil bacterial communities and its affecting factors with the method of high-throughput 454pyrosequencing technology, Soil samples were collected from four different arbor forests (Quercus liaotungensis, LDL; Biota orientalis, CB; Ptabulaeformis Carr; YS; Robinia pseucdoacacia, CH)), which represented the dominant communities for the forest vegetation ecosystem in the northwest of the Loess Plateau. The results showed that the structures of the microbial communities differed in terms of both the predominant phylum and the relative abundance of each phylum. At the phylum level, the dominant phyla were Proteobacteria, Actinobacteria, Acidobacteria, Chloroflexi and Planctomycetes. At the class level, the Actinobacteri, Alphaproteobacteria, Acidobacteria, Betaproteobacteria and Planctomycetacia were predominant. Compared with other arbor forests, the relative abundance of Acidobacteria and Bacteroidetes for LDL were the most abundant, while the relative abundance of Chloroflexi in YS and CB were more abundant than other vegetation types. Soil pH was significantly correlated with the relative abundance of Chloroflexi, and soil total phosphorus was significantly correlated with the relative abundance of Cyanobacteria, suggesting that soil total phosphorus was the main factor of affecting soil bacterial communities.
454 high-throughput sequencing;Ziwuling mountain;different arbor forests;soil bacteria;environment factors
S153.2
A
1000-6923(2016)11-3487-08
刘 洋(1991-),男,甘肃天水人,西北农林科技大学硕士研究生,主要从事生态环境工程研究.
2016-03-15
国家自然科学基金(41171226,41030532);水利部公益性行业科研专项经费项目(201501045);新世纪优秀人才支持计划(NCET-12-0479)
*责任作者, 副教授, ymhuang1971@nwsuaf.edu.cn
