顽痹清丸HPLC指纹图谱研究
2016-12-19罗石任王昭李洁
罗石任,王昭,李洁
河南省洛阳正骨医院河南省骨科医院,河南 郑州 450000
顽痹清丸HPLC指纹图谱研究
罗石任,王昭,李洁
河南省洛阳正骨医院河南省骨科医院,河南 郑州 450000
目的 建立顽痹清丸高效液相色谱(HPLC)指纹图谱评价体系,为顽痹清丸的质量控制提供依据。方法 以Agilent C18柱(4.6 mm×250 mm,5 μm)为色谱柱,以乙腈-0.1%磷酸水溶液为流动相梯度洗脱,流速1.0 mL/min,检测波长258 nm,进样量20 μL。对10批顽痹清丸样品进行测定,采用《中药色谱指纹图谱相似度评价系统(2004 A)》建立顽痹清丸的标准指纹谱,并计算各批样品的相似度。结果 所建立的顽痹清丸指纹图谱共确定20个共有峰,相似度不低于0.98,5个峰得到化学确认。结论 建立的HPLC指纹图谱方法稳定易行,重复性好,可用于顽痹清丸的质量评价。
顽痹清丸;高效液相色谱法;指纹图谱
顽痹清丸为本院医院制剂[豫药制字Z20120245(洛)],由忍冬藤、络石藤、桑枝、黄芩等14味药组成,具有清热除湿、祛风通络功效,适用于风湿热痹阻经络所致风湿性关节炎、类风湿关节炎、骨性关节炎等。中药指纹图谱能全面反映中药内在化学成分的种类与数量,利用指纹图谱相似度评价结果可直观分析不同批次制剂的质量差异,指导制剂生产过程中原药材的选择及生产工艺的优化[1-3]。本研究采用高效液相色谱法(HPLC)建立顽痹清丸指纹图谱,为全面控制其质量及进一步研究提供依据。
1 仪器与试药
Agilent 1100高效液相色谱仪(美国),XS205DU型分析天平(梅特勒-托利多有限公司),TP300型超声波提取器(天鹏电子新技术北京有限公司),HHS26s型数显恒温水浴锅(金坛市大地自动化仪器厂)。
顽痹清丸(批号20101109、20101220、20110105、20110214、20110321、20110510、20110615、20110723、20110903、20111020),河南省正骨研究所提供;对照品马钱苷(批号111640-201005)、黄芩苷(批号110715-201016)、蜕皮甾酮(批号110715-201016)、丹皮酚(批号110715-201016)、吴茱萸碱(批号110715-201016)均购自中国食品药品检定研究院;甲醇、乙腈为色谱纯,水为双蒸水,其余所用试剂均为分析纯。
2 方法与结果
2.1 对照品溶液制备
取马钱苷对照品、黄芩苷对照品、丹皮酚对照品、吴茱萸对照品、蜕皮甾酮对照品适量,精密称定,加甲醇制成浓度分别为40.50、138.8、124.3、68.5、118.9 μg/mL的对照品溶液。
2.2 供试品溶液制备
取各批次顽痹清丸适量,研细,取约2.5 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇40 mL,称定质量,超声处理(功率250 W,频率33 kHz)40 min,放至室温,再称定质量,用70%甲醇补足减失的质量,摇匀,过滤,滤液浓缩并定容至10 mL,用0.45 μm微孔滤膜过滤,作为供试品溶液。
2.3 色谱条件
色谱柱:Agilent C18柱(4.6 mm×250 mm,5 μm);以乙腈为流动相A,0.1%磷酸水溶液为流动相B,按表1进行梯度洗脱;记录时间为77 min;柱温:30 ℃;流速:1.0 mL/min;检测波长:258 nm;进样量:20 μL。

表1 流动相梯度洗脱程序(%)
2.4 方法学考察
2.4.1 重复性试验 分别精密称取同一批样品6份,按“2.2”项下方法制备供试品溶液,分别进样,记录指纹图谱。各共有峰的相对保留时间和相对峰面积积分值RSD均<3%,符合指纹图谱要求,表明该方法重复性良好。
2.4.2 精密度试验 精密吸取同一对照品溶液20 µL,按“2.2”项下色谱条件进样,连续进样5次,记录指纹图谱,结果显示,共有峰的相对保留时间和相对峰面积积分值的RSD均<3%,表明该方法精密度良好。
2.4.3 稳定性试验 取同一批供试品溶液,分别于0、4、6、8、12、24 h测定结果,记录指纹图谱,共有峰的相对保留时间和相对峰面积积分值RSD均<3%,符合指纹图谱要求,表明供试品溶液在24 h内稳定性良好。
2.5 顽痹清丸高效液相色谱指纹图谱
2.5.1 指纹图谱的建立 按“2.2”项下方法制备10批顽痹清丸样品,按“2.3”项下色谱条件进样,得到10批顽痹清丸HPLC指纹图谱,将其导入《中药色谱指纹图谱相似度评价系统(2004 A)》,进行色谱法匹配,结果见图1。所得指纹图谱共有模式见图2。以15号峰作为参照,计算各共有峰相对保留时间和相对峰面积,结果见表2、表3。
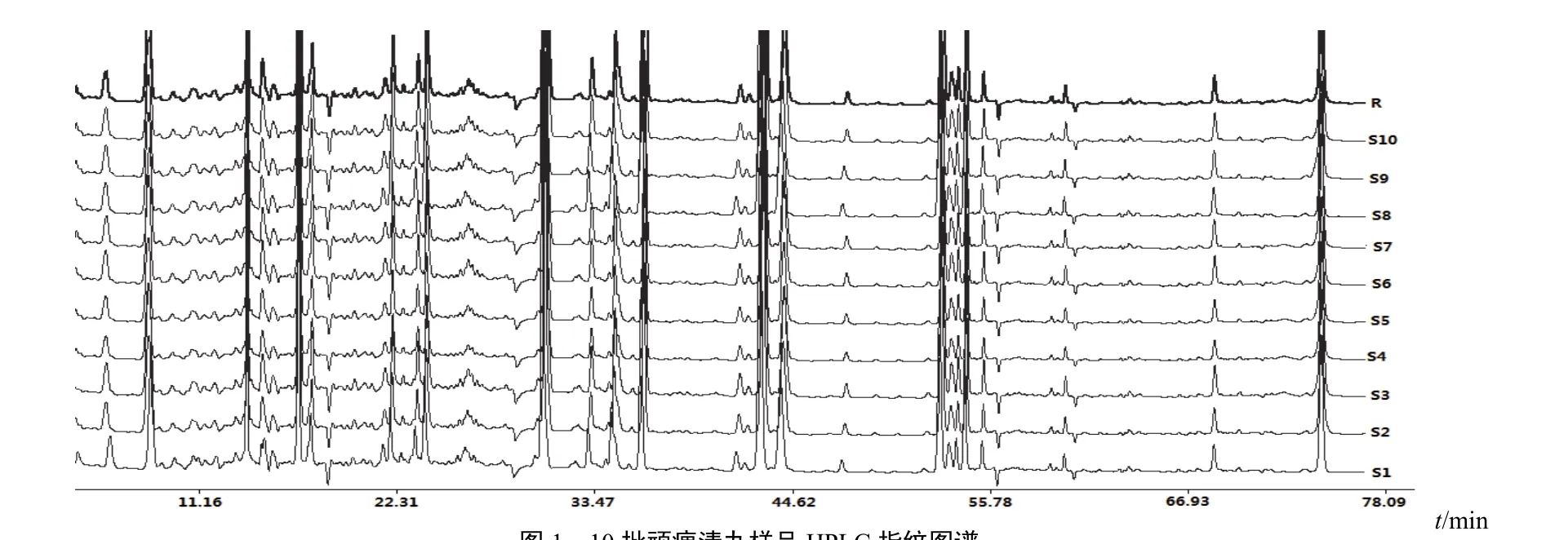
图110 批顽痹清丸样品HPLC指纹图谱

图2 顽痹清丸样品指纹图谱共有模式

表210 批顽痹清丸样品相对保留时间

表310 批顽痹清丸样品相对峰面积
2.5.2 指纹图谱相似度测定 采用国家药典委员会《中药色谱指纹图谱相似度评价系统(2004 A)》软件,采用相关系数法对10批顽痹清丸进行相似度计算,结果见表4。结果表明,10批样品相似度良好。
2.5.3 各峰归属的判断 通过与对照品保留时间进行对照,确认了其中5个指纹峰:5号峰为马钱苷,7号峰为蜕皮甾酮,10号峰为黄芩苷,15号峰为丹皮酚,19号峰为吴茱萸碱。对照品色谱图见图3。

批号 S1 S2 S3 S4 S5 S6 S7 S8 S9 S10 对照S1 1 0.987 0.996 0.994 0.996 0.997 0.995 0.993 0.994 0.994 0.997 S2 0.987 1 0.984 0.982 0.985 0.987 0.986 0.990 0.988 0.988 0.991 S3 0.996 0.984 1 0.991 0.992 0.999 0.997 0.994 0.995 0.995 0.997 S4 0.994 0.982 0.991 1 0.999 0.991 0.993 0.993 0.993 0.993 0.995 S5 0.996 0.985 0.992 0.999 1 0.992 0.994 0.995 0.995 0.995 0.997 S6 0.997 0.987 0.999 0.991 0.992 1 0.998 0.995 0.996 0.996 0.998 S7 0.995 0.986 0.997 0.993 0.994 0.998 1 0.997 0.998 0.998 0.999 S8 0.993 0.990 0.994 0.993 0.995 0.995 0.997 1 0.998 0.999 0.998 S9 0.994 0.988 0.995 0.993 0.995 0.996 0.998 0.998 1 0.988 0.999 S10 0.994 0.988 0.995 0.993 0.995 0.996 0.998 0.999 0.988 1 0.999对照 0.997 0.991 0.997 0.995 0.997 0.998 0.999 0.998 0.999 0.999 1
3 讨论
本试验考察了甲醇、50%甲醇、70%甲醇3种提取溶剂的提取效果[4-6],结果发现以50%甲醇作为提取溶剂的样品溶液所得峰数较多,但杂质也很多,很难使每个色谱峰完全分离,且所得色谱图特征不明显;甲醇为提取溶剂比70%甲醇和50%甲醇提取所得的样品溶液色谱峰少;70%甲醇作为提取溶剂的样品溶液所得的供试品峰数较多,且色谱峰分离度较好。因此选用70%甲醇作为提取溶剂。试验还考察了以70%甲醇为提取溶剂超声和水浴回流2种提取方法,结果发现超声提取所得色谱峰比回流所得色谱峰多,因此采用超声提取法作为供试品溶液的提取方法。
本试验曾采用甲醇-水、乙腈-水不同比例变化的流动相梯度洗脱系统进行洗脱[7-8],结果乙腈-水作为流动相时色谱峰分离效果比甲醇-水作为流动相峰的分离效果好,且基线较为平稳,但个别峰的分离效果不佳,在水相中加入0.1%磷酸进行调节,结果发现效果改善明显,各色谱峰的分离效果较好,最后确定流动相洗脱系统为乙腈-0.1%磷酸水溶液梯度洗脱[9]。试验曾采用236、258、265、274、280 nm波长进行测定[10],经比较后发现,258 nm波长处基线平稳、出峰稳定,故确定258 nm为检测波长。选用Agilent C18(4.6 mm×250 mm,5 μm)和Diamonsil C18(4.6 mm×250 mm,5 μm)色谱柱进行测定,结果表明,Agilent C18柱(4.6 mm×250 mm,5 μm)的分离效果较好,保留时间适宜,因此本试验选择Agilent C18柱为检测色谱柱。
[1] 程红,姚志红,戴毅,等.中药复方制剂仙灵骨葆胶囊HPLC指纹图谱研究[J].中国药学杂志,2013,48(10):772-776.
[2] 李文博,韩建平,高钧,等.养血清脑颗粒的高效液相色谱指纹图谱研究[J].分析化学研究简报,2011,39(3):387-391.
[3] 孙国祥,王建会.双黄连胶囊HPLC指纹图谱的不确定度和可靠度研究[J].时珍国医国药,2011,22(12):2831-2834.
[4] 唐云,刘汉清,唐海涛,等.蒲参胶囊的HPLC指纹图谱研究[J].中国实验方剂学杂志,2013,19(4):133-137.
[5] 刘伟,杨艳玲,刘乃强,等.柴胡口服液HPLC指纹图谱及柴胡皂苷b2的含量测定[J].中国实验方剂学杂志,2013,19(8):134-137.
[6] 张娴,徐英宏.乳腺增生丸高效液相色谱指纹图谱研究[J].医药导报, 2013,32(8):1093-1097.
[7] 李洁,罗石任,王秀真,等.桃仁膝康丸HPLC指纹图谱研究[J].中国中医药信息杂志,2013,20(10):59-62.
[8] 王文燕,赵强,张铁军,等.决明子的HPLC指纹图谱及模式识别研究[J].中草药,2009,40(10):1638-1641.
[9] 段天璇,于密密,刘春生,等.HPLC法同时测定甘草指纹图谱暨甘草苷、甘草酸含量[J].中成药,2006,28(2):161-165.
[10] 刘艳华,赵陆华,黄剑,等.丹参HPLC指纹图谱的研究[J].中国药科大学学报,2002,33(2):127-130.
Study on HPLC Fingerprint of Wanbiqing Pills
LUO Shi-ren, WANG Zhao, LI Jie (Luoyang
Orthopedic Hospital of Henan Province, Orthopedic Hospital of Henan Province, Zhengzhou 450000, China)
Objective To establish an evaluation system of HPLC fingerprint of Wanbiqing Pills; To provide references for the quality control of Wanbiqing Pills. Methods Wanbiqing Pills were analyzed with HPLC method. Analysis was performed on Agilent C18column (4.6 mm×250 mm, 5 μm) with the mobile phase of acetonitrile-0.1%H3PO4water gradient elution at the flow rate of 1.0 mL/min. The detection wavelength was 258 nm. Sample injection was 20 μL. Standard fingerprint of Wanbiqing Pills was established based on Similarity Evaluation System for Chromatographic Fingerprint of TCM (Version 2004 A). The similarity of Wanbiqing Pills from different batches was computed. Results A standard HPLC fingerprint procedure was developed for Wanbiqing Pills with 20 common peaks. The similarity of 10 batches of Wanbiqing Pills was not lower than 0.98. Five peaks received chemical confirmation. Conclusion The HPLC method is stable and feasible and with good repeatability, which can be used for the quality control of Wanbiqing Pills.
Wanbiqing Pills; HPLC; fingerprint
10.3969/j.issn.1005-5304.2016.01.023
R284.1
A
1005-5304(2016)01-0096-04
2015-03-20)
(
2015-04-10;编辑:陈静)
王昭,E-mail:18337960862@163.com
