Mg-6%Al-5%Pb阳极在3.5%NaCl溶液中的腐蚀行为
2016-12-15殷立勇王平安
殷立勇,周 威,王平安
(中国电子科技集团公司第十八研究所,天津300384)
Mg-6%Al-5%Pb阳极在3.5%NaCl溶液中的腐蚀行为
殷立勇,周威,王平安
(中国电子科技集团公司第十八研究所,天津300384)
采用熔炼铸造法制备Mg-6%Al-5%Pb(质量分数,下同)阳极材料,采用金相显微镜(OM)观察不同热处理状态下Mg-6%Al-5%Pb阳极材料的显微组织,采用X射线衍射仪(XRD)分析Mg-6%Al-5%Pb阳极材料的相结构,采用电化学方法和化学浸泡法研究Mg-6%Al-5%Pb阳极材料在3.5%NaCl溶液中的腐蚀行为,采用扫描电镜(SEM)观察浸泡不同时间后Mg-6%Al-5%Pb阳极材料的腐蚀表面形貌。结果表明:Mg-6%Al-5%Pb阳极材料在浸泡初期的腐蚀存在孕育期,孕育期内腐蚀速率较小。随着浸泡时间的延长,腐蚀逐渐进入稳态,腐蚀速率增大、趋于恒定。铸态试样的腐蚀以电偶腐蚀的形式从第二相Mg17Al12的周围开始,第二相能减小Mg-6%Al-5%Pb阳极材料的腐蚀速率,但局部腐蚀严重。400℃固溶24 h后腐蚀以点蚀的形式从晶界处开始,腐蚀速率相比铸态试样大,但腐蚀相对铸态试样均匀。
镁阳极;腐蚀速率;耐蚀性;析氢;电化学
镁合金阳极材料的海水激活动力电池具有电化学活性高、电压范围广、能量密度大、未被激活时储存时间长[1-3]等特点,广泛应用于海底声音测试装置、潜水艇、警告浮标、球状电池、空间飞行器和紧急救生设备等领域[4]。海水电池依靠阳极金属材料在海水中的腐蚀溶解提供阳极放电电流,而阴极则主要依靠海水中的溶解氧在惰性的气体电极上进行还原反应提供阴极电流。海水电池最突出的特点是不需要携带电解质,可以在需要的时候利用天然海水形成电解液,在运输过程中绝对安全[5]。但镁合金阳极材料存在难加工、自腐蚀速率大且电流效率低等缺点[6-8],目前解决此类问题的方法是改善热处理制度和添加适量的合金元素。AP65是镁合金阳极中的一种,其名义成分为Mg-6%Al-5%Pb(质量分数)。Al影响镁阳极的耐蚀性,其效果取决于Al的含量和第二相的分布[9-12]。适量的Pb可以增强镁阳极的耐蚀性[13]。目前国内外关于Mg-6%Al-5%Pb阳极材料腐蚀行为的报道较少。本文作者研究铸态和固溶态Mg-6%Al-5%Pb阳极材料在3.5%NaCl溶液中的腐蚀行为,目的在于比较不同热处理状态下Mg-6%Al-5%Pb阳极材料的耐腐蚀性能并探讨各状态下Mg-6%Al-5%Pb阳极材料的腐蚀机制。
1 实验
采用感应熔炼法制备Mg-6%Al-5%Pb(质量分数,下同)阳极材料,将Mg、Al、Pb等以纯金属(纯度99.99%)放入高纯石墨坩埚,750℃熔炼,充氩气保护。金属熔体在氩气保护下于水冷铁模浇铸。所得试样于400℃固溶24 h,水淬。采用原子吸收光谱分析Mg-6%Al-5%Pb阳极材料的化学成分,列于表1。采用POLVAR-MET宽视野大型金相显微镜观察经打磨、抛光后各试样的显微组织。采用D/Max2500衍射仪分析铸态和固溶态Mg-6%Al-5%Pb阳极材料的相结构[扫描速度为1.2(°)/min,扫描范围为10°~80°,Cu靶]。
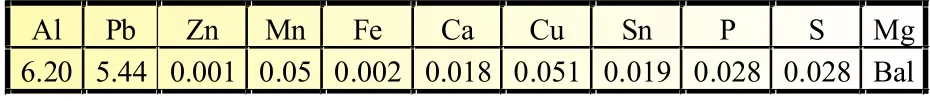
表1 Mg-6%AI-5%Pb阳极的化学成分(质量分数,%)
采用动电位极化扫描法和交流阻抗法测定试样的耐腐蚀性能。将试样用同型号SiC砂纸打磨去除表面氧化层,保留其经打磨的工作面并使其呈10mm×10mm的矩形。然后用铜导线捆绑样品,非工作面用环氧树脂密封。电化学仪器为IM6ex,采用三电极体系进行测量。工作电极为Mg-6%Al-5% Pb阳极,辅助电极为铂电极,参比电极为饱和甘汞电极。实验温度为25℃,电解液为3.5%(质量分数)的NaCl中性溶液。动电位极化扫描法的扫描速度为2 mV/s,电压范围为开路电位±0.8 V;交流阻抗法的测试电位为开路电位,频率范围为100 kHz~0.05 Hz,电压振幅为5 mV。各试样的电化学测试在试样浸入溶液后立即进行。
采用化学浸泡法研究试样的腐蚀行为。用排水法收集试样浸泡过程中析出的氢气。将试样用同型号SiC砂纸打磨去除表面氧化层,保留其经打磨的工作面并使其呈10mm×10mm的矩形,非工作面用环氧树脂密封。实验温度为25℃,电解液为3.5%的NaCl中性溶液,浸泡时间为72 h,测定析出氢气的体积随时间的变化规律,计算出试样浸泡6和72 h的平均析氢速率。
采用JSM-5600Lv扫描电镜观察试样在3.5%的NaCl溶液中浸泡6和48 h的腐蚀表面形貌以及浸泡48 h后的腐蚀截面。用200 g/L CrO3+10 g/L AgNO3溶液清除各试样的腐蚀产物,并将清除腐蚀产物的试样在无水乙醇中超声波清洗5 min,干燥并观察试样的腐蚀形貌。
2 结果与分析
2.1Mg-6%Al-5%Pb阳极材料不同状态下的显微组织和相结构
图1所示为铸态和固溶态Mg-6%Al-5%Pb阳极材料的金相照片。从图1(a)可以看出,铸态试样中除镁基体外还存在大量的第二相。根据图1(b)放大的金相照片可知,第二相主要在晶界处不连续分布,晶内第二相较少。根据图2(a)铸态试样的XRD图谱可知,该第二相为Mg17Al12,基体为α-Mg。经400℃固溶24 h后,第二相溶解,试样成为单相固溶体组织[图1(c)]。根据图2(b)固溶态试样的XRD图谱可知,固溶体组织中仅存在α-Mg相。

图1 Mg-6%Al-5%Pb阳极材料的金相照片
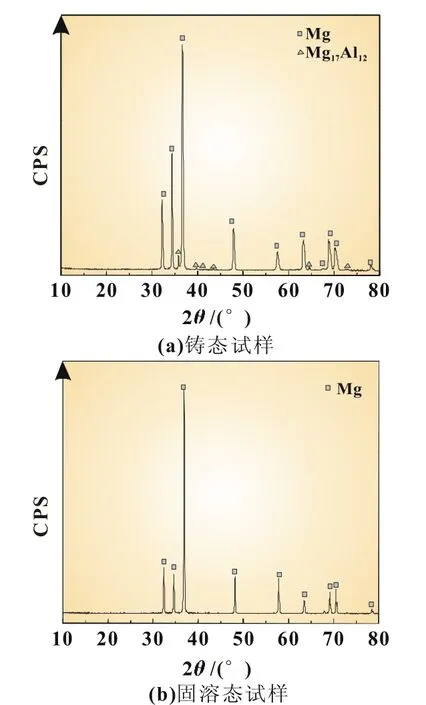
图2 Mg-6%Al-5%Pb阳极材料的XRD图谱
2.2Mg-6%Al-5%Pb阳极材料不同状态下的动电位极化扫描曲线
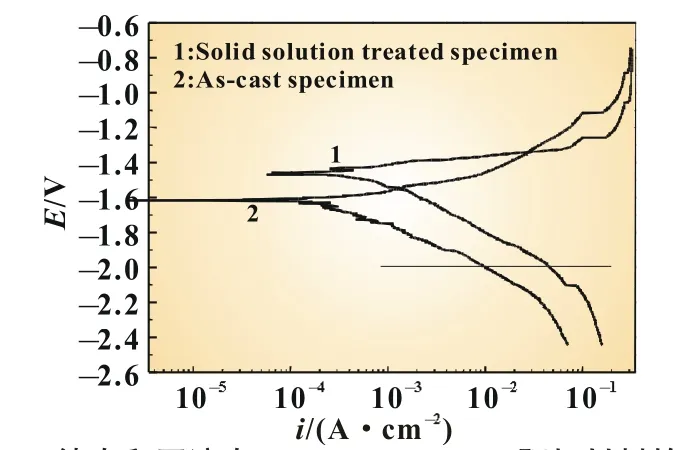
图3 铸态和固溶态Mg-6%Al-5%Pb阳极材料的动电位极化扫描曲线

表2 Mg-6%AI-5%Pb阳极材料不同状态下的腐蚀电位(E )和腐蚀电流密度(i )
图3所示为铸态和固溶态Mg-6%Al-5%Pb阳极材料的动电位极化扫描曲线。各试样的阳极支和阴极支不对称,阳极支电流密度随电位增加的速率高于阴极支。一般来说,阴极支的电流主要受析氢控制,阳极支的电流则主要受金属的阳极溶解控制[14]。在动电位极化扫描过程中,黑色腐蚀产物不断从试样表面剥落,反应中没有钝化行为出现。根据动电位极化扫描曲线计算出各试样的腐蚀电位和腐蚀电流密度列于表2。从表2可以看出,固溶态试样的腐蚀电位为-1.428 V(vs.SCE),高于铸态试样的腐蚀电位-1.598 V(vs.SCE)。固溶态试样的腐蚀电流密度为0.531 mA/cm2,大于铸态试样的腐蚀电流密度0.225 mA/cm2。镁合金阳极材料中普遍存在电偶腐蚀,第二相比镁基体具有更高的电极电位而充当阴极[6],其分布和数量对电化学腐蚀行为有很大影响。当第二相的数量较少时,主要作为阴极相与镁基体形成腐蚀电偶加速镁基体的腐蚀;当第二相的数量较多时,则主要作为腐蚀屏障抑制镁基体的腐蚀[15]。结合图1的金相照片和图2的XRD图谱可知,铸态试样晶界处存在不连续分布的第二相Mg17Al12,固溶态试样中Mg17Al12相数量稀少。虽然第二相Mg17Al12相对镁基体为阴极相,但铸态试样中Mg17Al12相数量较多,能作为腐蚀屏障抑制镁基体的腐蚀[16]。因此,Mg-6%Al-5%Pb阳极材料铸态试样的腐蚀电流密度小于固溶态试样。根据图3可知,在同一阴极电位下(横线所示),铸态试样的阴极电流密度低于固溶态试样,表明该状态下的第二相Mg17Al12能有效抑制阴极反应过程中氢气的析出。腐蚀电流密度可以采用式(1)换算成腐蚀速率[17]:

式中:Pi为腐蚀速率,mm/a;icorr为腐蚀电流密度,mA/cm2。根据公式(1)计算出Mg-6%Al-5%Pb阳极各试样的腐蚀速率,列于表3。由表3可知,铸态和固溶态Mg-6%Al-5%Pb阳极根据析氢速率计算出的腐蚀速率与腐蚀电流密度计算出的腐蚀速率呈现相同的变化趋势。
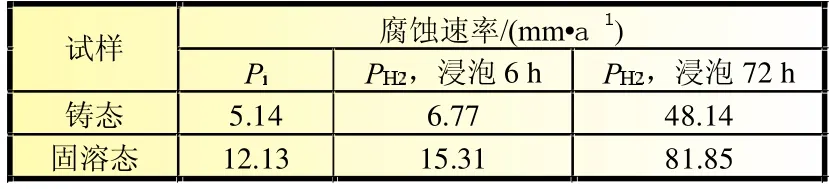
表3 根据腐蚀电流密度和平均析氢速率计算所得的Mg-6%AI-5%Pb阳极材料不同状态下的腐蚀速率
2.3Mg-6%Al-5%Pb阳极材料不同状态下的电化学阻抗谱
图4所示为铸态和固溶态Mg-6%Al-5%Pb阳极材料的电化学阻抗谱。根据图4(a)所示的Nyquist图可知,铸态试样在整个频率范围内仅存在一个容抗弧,该容抗弧与双电层电容Cdl和电荷转移电阻Rt有关,表明铸态试样的电化学过程仅受电极电位控制。固溶态试样除了在高频区存在一个与双电层电容Cdl和电荷转移电阻Rt有关的容抗弧外,在低频区也存在一个容抗弧,该容抗弧与沉积在试样表面的腐蚀产物膜有关,其电容和电阻分别用Cf和Rf表示。因此固溶态试样的电化学过程除了受电极电位控制外,还受沉积在表面的腐蚀产物膜控制。根据图4(b)所示的Bode图可知,铸态试样的阻抗模值在整个频率范围内都大于固溶态试样,表明铸态试样与固溶态相比具有更强的耐蚀性,与表2所列的腐蚀电流密度具有相同的规律。根据电化学阻抗谱拟合出各试样的电化学元件参数值列于表4,并做出各试样的等效电路图(见图5)。在等效电路图中,Rs表示试样表面与鲁金毛细管之间的溶液电阻。由于弥散效应的存在,采用常相位角元件CPEdl代替双电层电容Cdl,该元件与电荷转移电阻Rt并联。从表4可以看出,固溶态试样的电荷转移电阻小于铸态试样,表明固溶态试样与铸态相比具有更强的电化学活性。因此固溶态试样在NaCl溶液中浸泡时有更多的Mg2+离子溶解到溶液中,当Mg2+离子的浓度超过某一极限值时,将以Mg(OH)2的形式在试样表面沉积[17],导致固溶态试样的Nyquist图在低频区出现容抗弧[18]。

图4 铸态和固溶态Mg-6%Al-5%Pb阳极材料的电化学阻抗谱

表4 根据电化学阻抗谱拟合出的Mg-6%AI-5%Pb阳极材料不同状态下的电化学参数
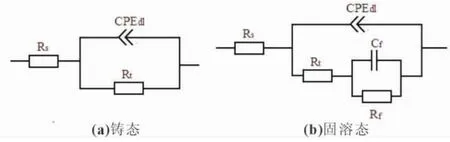
图5 不同状态下Mg-6%Al-5%Pb阳极材料的等效电路图
2.4Mg-6%Al-5%Pb阳极材料不同状态下的析氢行为
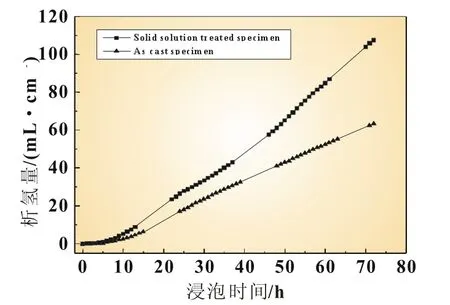
图6 铸态和固溶态Mg-6%Al-5%Pb阳极材料的析氢体积随浸泡时间的变化曲线
图6所示为铸态和固溶态Mg-6%Al-5%Pb阳极材料在3.5%NaCl溶液中浸泡时析出氢气的体积随浸泡时间的变化曲线。根据图6可知,在浸泡的开端各试样的腐蚀存在孕育期,在孕育期内析氢速率(即曲线的斜率)较小。随着浸泡时间的延长,腐蚀逐渐进入稳态,析氢速率增大直至趋于恒定。因为在浸泡的初始阶段试样表面只有少数区域被腐蚀,氢气在这些区域析出,因此析氢速率较小。随着浸泡时间的延长,腐蚀区域逐渐增大,直至蔓延到试样的整个表面,因此析氢速率逐渐增大直至趋于恒定。在任何一个浸泡时间点上,铸态试样的析氢体积均小于固溶态试样,表明铸态试样相比固溶态有较强的耐蚀性,这与铸态试样中存在的第二相Mg17Al12有关,该第二相能抑制氢气的析出。
各试样的平均析氢速率可以采用式(2)计算:

式中:v(H2)为平均析氢速率,mL/(cm2·h);V(VH2)为析氢体积,mL;t为浸泡时间,h;S为试样表面积,cm2。根据公式(2)计算出Mg-6%Al-5%Pb阳极各试样浸泡6和72 h的平均析氢速率,列于表5。
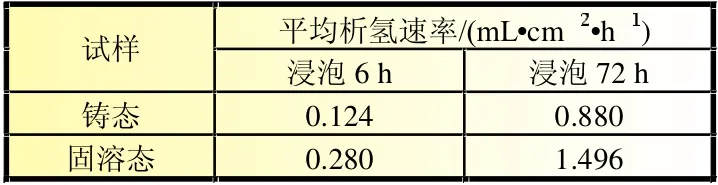
表5 Mg-6%AI-5%Pb阳极材料不同状态下浸泡6和72 h的平均析氢速率
平均析氢速率v(H2)可以采用式(3)换算成腐蚀速率[17]:

式中:PH2为腐蚀速率,mm/a。根据公式(3)计算出Mg-6%Al-5% Pb阳极各试样浸泡6和72 h的腐蚀速率,列于表3。由表3可知,铸态试样的腐蚀速率小于固溶态试样。各试样根据腐蚀电流密度计算出的腐蚀速率接近于根据浸泡6 h的平均析氢速率计算出的腐蚀速率,但小于根据浸泡72 h的平均析氢速率计算出的腐蚀速率。因此采用动电位极化扫描法测得的腐蚀速率仅能反映试样在腐蚀孕育期内的耐蚀性,而不能反映试样经长时间浸泡后进入腐蚀稳态的耐蚀性。
2.5Mg-6%Al-5%Pb阳极材料不同状态下的腐蚀形貌
图7所示为不同状态下Mg-6%Al-5%Pb阳极材料在3.5%NaCl溶液中浸泡6 h后腐蚀表面形貌的二次电子像。铸态试样在浸泡6 h后第二相周围的镁基体被腐蚀,表明铸态试样的腐蚀类型为电偶腐蚀[图7(a)],该腐蚀主要从第二相周围的镁基体开始。从图7(b)可以看出,固溶态试样在浸泡6 h后表面出现大量点蚀孔,表明固溶态试样的腐蚀类型为点蚀。根据图7(c)放大的二次电子像可知,点蚀孔主要分布在晶界,表明点蚀主要从晶界处开始。图8所示为各试样浸泡48 h后腐蚀表面形貌的二次电子像。从图8(b)可以看出,固溶态试样在浸泡48 h后整个表面都已被腐蚀,表明点蚀孔的数量随浸泡时间的延长而增多,且点蚀孔增大,直至蔓延到整个试样表面。浸泡48 h后固溶态试样的晶界处仍存在大量点蚀孔,表明试样经长时间浸泡后腐蚀不断往下挖深,同样以点蚀的形式从晶界处开始。铸态试样在浸泡48 h后整个表面也已被腐蚀,第二相Mg17Al12依然可见[图8(a)]。相比固溶态,铸态试样的腐蚀表面凹凸不平,第二相周围的镁基体相比其他区域腐蚀得更严重,表明铸态试样经长时间浸泡后腐蚀不断往下挖深,同样以电偶腐蚀的形式从第二相周围开始。图9所示为各试样浸泡48 h后腐蚀截面的低倍背散射像。从图9(b)可以看出,固溶态试样浸泡48 h后腐蚀均匀,而铸态试样则存在严重的局部腐蚀[图9(a)]。固溶态试样由于成分均匀,在浸泡过程中试样均匀溶解,且活性较强,适合用于大功率海水电池阳极材料;铸态试样尽管腐蚀速率小于固溶态试样,但由于第二相的存在,导致局部腐蚀严重且活性较差,不适合用于大功率海水电池阳极材料。
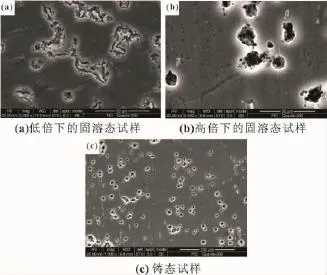
图7 Mg-6%Al-5%Pb阳极材料在3.5%NaCl溶液中浸泡6 h后腐蚀表面形貌的二次电子像
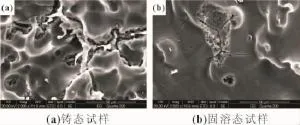
图8 Mg-6%Al-5%Pb阳极材料在3.5%NaCl溶液中浸泡48 h后腐蚀表面形貌的二次电子像

图9 Mg-6%Al-5%Pb阳极材料在3.5%NaCl溶液中浸泡48 h后腐蚀截面的背散射像
3 结论
(1)Mg-6%Al-5%Pb阳极材料在浸泡的开端腐蚀存在孕育期,在孕育期内腐蚀速率较小。随着浸泡时间的延长,腐蚀逐渐进入稳态,腐蚀速率增大且趋于恒定。根据动电位极化扫描法测得的Mg-6%Al-5%Pb阳极材料腐蚀速率仅能反映试样在腐蚀孕育期内的耐蚀性,而不能反映试样经长时间浸泡后进入腐蚀稳态的耐蚀性。
(2)Mg-6%Al-5%Pb阳极材料铸态试样的腐蚀以电偶腐蚀的形式从第二相Mg17Al12的周围开始且第二相能减小Mg-6%Al-5%Pb阳极材料的腐蚀速率,但局部腐蚀严重。浸泡48 h后腐蚀蔓延至整个表面且不断往下挖深,同样以电偶腐蚀的形式从第二相的周围开始;固溶态试样的腐蚀以点蚀的形式从晶界处开始且腐蚀速率大于铸态试样,但腐蚀相比铸态试样均匀。浸泡48 h后腐蚀蔓延至整个表面且不断往下挖深,同样以点蚀的形式从晶界处开始。
[1]MEDEIROS M G,DOW E G.Magnesium-solution phase catholyte seawater electrochemical system[J].Journal of Power Sources, 1999,80(1/2):78-82.
[2]RENUKA R.Influence of allotropic modifications of sulphur on the cell voltage in Mg-CuI(S)seawater activated battery[J].Materials Chemistry and Physics,1999,59(1):42-48.
[3]MEDEIROS M G,BESSETTE R R,DESCHENES C M,et al.Optimization of the magnesium-solution phase catholyte semi-fuel cell for long duration testing[J].Journal of Power Sources,2001,96 (1):236-239.
[4]RENUKA R.AgCl and Ag2S as additives to CuI in Mg-CuI seawater activated batteries[J].Journal of Applied Electrochemistry, 1997,27(12):1394-1397.
[5]KIM J G,JOO J H,KOO S J.Development of high-driving potential and high-efficiency Mg-based sacrificial anodes for cathodic protection[J].Journal of Materials Science Letters,2000,19(6): 477-479.
[6]王乃光,王日初,余琨,等.合金化和热处理对镁合金阳极材料组织及性能的影响[J].中国有色金属学报,2009,19(1):38-43.
[7]马正青,曹军纪.海水介质中高活性镁合金负极的电化学性能[J].材料保护,2002,35(12):16-18.
[8]FLAMINI D O,SAIDMAN S B,BESSONE J B.Aluminium activation produced by gallium[J].Corrosion Science,2006,48(6): 1413-1425.
[9]ZHAO M C,SCHMUTZ P,BRUNNER S,et al.An exploratory study of the corrosion of Mg alloys during interrupted salt spray testing[J].Corrosion Science,2009,51:1277-1292.
[10]GU X N,ZHENG Y F,CHENG Y,et al.In vitro corrosion and biocompatibility of binary magnesium alloys[J].Biomaterials, 2009,30:484-498.
[11]PARDO A,MERION M C,COY A E,et al.Influence of microstructrue and composition on the corrosion behaviour of Mg/Al alloys in chloride media[J].Electrochimica Acta,2008,53:7890-7902.
[12]ZHAO M C,LIU M,SONG G L,et al.Influence of the β-phase morphology on the corrosion of the Mg alloy AZ91[J].Corrosion Science,2008,50:1939-1953.
[13]CANDAN S,UNAL M,TURKMEN M,et al.Improvement of mechanical and corrosion properties of magnesium alloy by lead addition[J].Material Science and Engineering A,2009,501:115-118.
[14]TAMAR Y,MANDLER D.Corrosion inhibition of magnesium by combined zirconia silica sol-gel films[J].Electrochimica Acta,2008,53:5118-5127.
[15]SONG G L,ATRENS A,DARGUSCH M.Influence of microstructure on the corrosion of diecast AZ91D[J].Corrosion Science,1999,41:249-273.
[16]冯艳.Mg-Hg-Ga阳极材料合金设计及性能优化[D].长沙:中南大学,2009.
[17]ZHAO M C,LIU M,SONG G L,et al.Influence of pH and chloride ion concentration on the corrosion of Mg alloy ZE41[J]. Corrosion Science,2008,50:3168-3178.
[18]曹楚南.腐蚀电化学原理[M].北京:化学工业出版社,2008.
Corrosion behavior of Mg-6%Al-5%Pb anode in 3.5%NaCl solution
YIN Li-yong,ZHOU Wei,WANG Ping-an
(Tianjin Institute of Power Sources,Tianjing 300384,China)
Mg-6%Al-5%Pb(mass fraction)anode materials were prepared by melting and casting.The microstructures of Mg-6%Al-5%Pb anodes under different heat treatment conditions were observed by optical microscopy(OM).The phase structures of Mg-6%Al-5%Pb anodes were analyzed by X-ray diffraction(XRD)and the corrosion behaviors of Mg-6%Al-5%Pb anodes in 3.5%NaCl solution were studied by electrochemical measurements and immersion tests.The corroded surfaces of Mg-6%Al-5%Pb anodes after various periods of immersion were observed by scanning electronic microscopy(SEM).The results show that there is an incubation period at the corrosion onset,with the low corrosion rate.Thereafter the corrosion rate increases as the increase of immersion time and gradually comes into steady state.For as-cast specimens,the galvanic corrosion initiates surrounding the second phases(Mg17Al12).The second phases can reduce the corrosion rate of the Mg-6%Al-5%Pb anode,while accelerate the local corrosion.After solid solution treatment at 400℃for 24 h,the corrosion of the Mg-6%Al-5%Pb anode initiates along the grain boundaries in the form of pitting corrosion and the corrosion rate is higher than that of as-cast specimen,but the corrosion develops homogeneously on the specimen surface.
magnesium anode;corrosion rate;corrosion resistance;hydrogen evolution;electrochemistry
TM 911
A
1002-087 X(2016)10-1997-05
2016-03-15
国家自然科学基金(51401243);湖南省科技计划项目(2015JC3004)
殷立勇(1977—),男,河北省人,高级工程师,主要研究方向为化学电源。E-mail:fengyanmse@csu.edu.cn
