酸处理石墨化碳载体对燃料电池催化剂性能的影响
2016-12-14闫海旭杨美妮浦鸿汀
闫海旭,杨美妮,曾 浩,浦鸿汀,林 瑞
(1.同济大学新能源汽车工程中心,上海201804;2.同济大学汽车学院,上海201804;3.同济大学材料科学与工程学院,上海201804)
酸处理石墨化碳载体对燃料电池催化剂性能的影响
闫海旭1,2,杨美妮1,2,曾浩1,2,浦鸿汀3,林瑞1,2
(1.同济大学新能源汽车工程中心,上海201804;2.同济大学汽车学院,上海201804;3.同济大学材料科学与工程学院,上海201804)
通过1700℃高温处理XC⁃72CB得到石墨化碳黑(GCB),并采用酸处理对GCB碳载体进行官能团修饰.透射电子显微镜(TEM)、X射线粉末衍射(XRD)和拉曼光谱的结果显示,酸处理后GCB的石墨化程度增加;N2吸附⁃脱附结果证明GCB比表面积减小,微孔数量减少;热重分析结果表明,GCB热稳定性增强;红外光谱和拉曼光谱结果显示,GCB表面引入了含氧官能团,并同时保持了GCB的有序化结构.采用循环伏安(CV)法和线性扫描伏安(LSV)法测试了不同预处理后催化剂的电化学性能,表明其电化学活性表面积(ECSA,75.25 m2/g)和质量比活性(MA,0.093 A/mg)均高于商业Pt/C(JM)催化剂.TEM结果表明,使用经过浓硫酸和浓硝酸混合酸处理的GCB(简称OGCB)作为载体得到的Pt/OGCB平均粒径为2.28 nm,略小于商业Pt/C(JM)催化剂(约2.5 nm);经5000周电化学循环伏安测试后,Pt/OGCB的电化学活性表面积衰减17.3%,质量比活性衰减29.5%,而Pt/C(JM)的ECSA衰减达到25.1%,MA衰减达到42.5%.
质子交换膜燃料电池;石墨化碳黑;酸处理;耐久性
质子交换膜燃料电池(PEMFC)具有能量转换效率高和能量密度高的优点,在车用燃料电池中已得到广泛应用[1,2].PEMFC的关键部分为膜电极(MEA),其中燃料电池催化剂的催化效率决定燃料电池的效率[3,4].PEMFC在酸性环境及车用频繁启停工况下可能发生碳腐蚀,进而使燃料电池的性能降低[4~6].因此,提高催化剂载体的耐久性至关重要[7].催化剂载体不仅对催化剂活性组分起到支撑和分散作用,同时还影响着催化剂的传质、传热及稳定性[8,9],提高贵金属在载体上的分散性可以提高贵金属的利用率[10,11].碳载体材料应具有较高的比表面积、高度的石墨化结构和适当的孔结构分布[12,13].
石墨化是碳材料由无定形及乱层结构向三维石墨结构转化的相变过程,sp2杂化使其具有较高的导电性、导热性和较高的化学稳定性及耐腐蚀性.商业的Vulcan XC⁃72碳黑(XC⁃72CB)石墨化程度较低,具有大量的微孔结构和表面缺陷[14],这种结构虽然有利于Pt的附着,但也是碳氧化腐蚀最容易发生的地方[15].碳黑经过高温处理后,大量的微孔发生塌陷,提高了碳材料的有序化程度[16];但过多微孔的塌陷也会造成比表面积的降低,降低了催化剂的负载.Hayashi等[17]研究发现,通过1600℃热处理后的XC⁃72 CB碳载体具有较高的石墨化程度,在0.6~1.0 V下经60000周的电化学循环耐久性测试后,其电化学活性表面积(ECSA)衰减较慢.虽然石墨化碳黑(GCB)作为载体具有很高的热稳定性和耐久性,但经过高温石墨化后的载体比表面积降低,且GCB的化学惰性表面使Pt难以负载并分散,负载的Pt颗粒移动碰撞发生团聚,粒径增大,导致初始ECSA减小[18].为此,Yano等[19]利用纳米胶囊法制备了Pt/GCB催化剂.该方法在Pt表面引入表面活性分子,形成的纳米胶囊微胶粒发生了排斥反应,从而使Pt纳米颗粒的聚集受到抑制,Pt纳米颗粒均匀分散于载体表面[20].此外,合理的颗粒间距阻止了Pt颗粒在“电化学奥斯特瓦尔德熟化”过程中长大,从而提高了催化剂的耐久性[21].Kim等[22]通过使石墨化碳黑表面发生重氮反应,在载体表面嫁接了三氟甲基苯硼酸官能团.原位嫁接层阻止了Pt的迁移,防止了其集聚结块.结果显示,重氮修饰的催化剂表现出了较高的电化学活性和稳定性.虽然通过引入表面活性分子或官能团修饰GCB可提高其负载能力,但这些制备方法不易操作且表面活性剂等原料较贵,难以满足大规模应用的需求.
本文在1700℃高温石墨化处理XC⁃72 CB得到GCB的基础上,将酸处理得到的含氧官能团修饰的GCB材料作为载体,采用脉冲微波法制备Pt/GCB催化剂,并测试了该催化剂的电化学性能.
1 实验部分
1.1 试剂与仪器
XC⁃72 CB(美国Cabot公司);质子交换膜(Nafion212)和Nafion溶液(质量分数5%,美国Dupon公司);Pt/C催化剂(Pt质量分数为20%,简写为Pt/C(JM),美国Johnson Matthy公司);高氯酸(质量分数70%,美国BE公司);氯铂酸、盐酸、浓硝酸和浓硫酸(分析纯,国药集团上海化学试剂公司);氮气和氧气(纯度≥99.99%,上海成功气体有限公司);氢气(纯度≥99.99%,上海宝氢气体工业有限公司).DZF⁃6020型真空干燥箱(上海精宏实验设备有限公司);Ad01型石墨化炉(株洲金瑞中高频设备有限公司);SHZ⁃DⅢ型真空冷冻干燥机和HOS⁃1型恒温磁力搅拌器(上海东玺制冷仪器设备公司);SCQ⁃70型超声清洗器(上海声彦超声波仪器有限公司);CHI 660型电化学工作站(上海辰华仪器公司);自制单电池测试平台;AL104⁃IC型电子天平(梅特勒⁃托利多仪器有限公司);DKB⁃501A型恒温水槽(上海申贤恒温设备厂);AFMS⁃LXF型旋转圆盘电极(RDE,美国PINE设备公司);ASAP2020型比表面及孔隙分析仪(美国Micrometrics公司);STA449C型热重⁃差热分析仪(德国Netzsch公司);InVia型激光拉曼光谱仪(英国Renishow公司);S⁃4800型红外光谱分析仪和S⁃4800型扫描电子显微镜(日本日立公司);PHI 5000C ESCA System型X射线光电子能谱分析仪(美国PHI公司);JEM⁃2010型透射电子显微镜(日本电子光学公司);D8 ADVANCE X型X射线衍射仪(德国Bruker公司);Axis Ultra DLD型X射线光电子能仪(英国Kratos公司).
1.2 GCB和催化剂的制备
1.2.1 GCB的制备 称取一定量XC⁃72 CB在氩气气氛下进行高温热处理,升温速率为5℃/min,热处理温度为1700℃,恒温处理1.5 h后冷却至室温,得到GCB.
1.2.2 GCB的预处理 采用不同的酸处理GCB.步骤如下:首先,称取3份0.1 g GCB,分别置于8 mL的浓硝酸(HNO3)、8 mL浓硫酸(H2SO4)和8 mL浓硫酸与浓硝酸(体积比为3∶1)的混合溶液中,搅拌均匀,然后,在120℃下回流加热8 h,冷却后离心,过滤;最后,在80℃恒温箱中干燥8 h.得到的材料分别标记为HNO3⁃GCB,H2SO4⁃GCB和H2SO4+HNO3⁃GCB(简称OGCB).
1.2.3 以GCB为载体的Pt催化剂的制备 采用脉冲微波辅助化学还原法合成催化剂.分别称取0.015 g GCB,HNO3⁃GCB,H2SO4⁃GCB和OGCB于烧杯中,分别加入4 mL乙二醇和2 mL丙酮的混合溶液,搅拌与超声交替进行1 h,再分别加入1 mL 0.019 mol/L H2PtCl6的乙二醇溶液,在常温下搅拌及超声各0.5 h后,用1 mol/L的氢氧化钠乙二醇溶液将混合物pH值调节至约11.待反应物分散均匀后,将混合液置于N2气氛围的微波炉中进行脉冲反应,反应条件:微波功率为2 kW,微波炉工作时间为12 s/次,微波炉弛豫时间为180 s/次,工作及弛豫的重复次数为4次.待温度降到30℃以下后,取出溶液并用稀盐酸调节pH值至3以下,静置12 h后过滤,最后真空冷冻干燥5 h.所得催化剂依次标记为Pt/GCB,Pt/HNO3⁃GCB,Pt/H2SO4⁃GCB和Pt/OGCB.
1.3 电化学测试
催化剂的性能使用美国PINE公司的三电极体系旋转圆盘电极在CHI660电化学测试系统上进行测试.采用可逆氢电极(RHE)为参比电极,铂丝为对电极,工作电极为涂有催化剂层的玻碳电极.在0.1 mol/L HClO4的电解质溶液中于室温下进行循环伏安(CV)与线性扫描伏安(LSV)测试.
将2 mg催化剂与1 mL甲醇/Nafion溶液(甲醇与Nafion质量比为30∶1)超声混合均匀配制成墨水状浆液.再移取10 μL墨汁涂到玻碳电极(S=0.283 cm2)上并在空气中干燥,制备成工作电极.CV测试时,通N2气30 min至饱和,扫描电位范围0.05~1.15 V,扫描速度0.05 V/s.LSV测试时,通O2气30 min至饱和,扫描电位范围0~1.2 V,扫描速度为0.005 V/s,圆盘转速为1600 r/min.
随后进行了5000周电化学循环伏安测试,测试范围为0.05~1.15 V,扫描速度为50 mV/s.
1.4 单电池性能测试
将Pt/OGCB作为阴极催化剂,Pt/C(JM)催化剂作为阳极催化剂,制备有效面积为50 cm2的MEA,并组装成单电池以评价自制催化剂的单电池性能.其中,阴极催化剂Pt负载量为0.4 mg/cm2,阳极催化剂的Pt负载量为0.2 mg/cm2.同时,以相同Pt负载量的Pt/C(JM)催化剂作为阴、阳极催化剂制备另一个MEA作为对比.把制备好的MEA组装到单电池的测试平台上进行测试,阳极以纯氢气为燃料,阴极以空气为氧化剂,氢气和空气的体积比为1.28∶3.2,空气和氢气相对湿度均为80%,进气压力均为20 kPa,使用蛇形金属流场板.
2 结果与讨论
2.1 GCB的表征
2.1.1 TEM表征 图1(A)是GCB的透射电子显微镜(TEM)照片.可见,碳材料表层出现了明显的石墨化片层结构,表明高温热处理提高了碳黑的有序化程度.图1(B)是GCB的选区电子衍射(SAED)谱图.图中可见强的(002)晶面的衍射弧,同心圆环的出现是因为入射束照射的样品区域内存在大量取向杂乱的细小晶粒.通过计算可得d(002)=0.3450 nm,接近石墨的d(002)=0.3354 nm,表明经过1700℃处理后载体已具备较高的石墨化程度.

Fig.1 TEM image(A)and SAED pattern(B)of GCB
2.1.2 X射线粉末衍射表征 图2是XC⁃72 CB经1700℃处理前后的X射线粉末衍射(XRD)谱图. 2θ=25.5°和43.2°处分别是C(002)和C(100)晶面的衍射峰.经1700℃热处理后,(002)和(100)晶面的衍射峰变得更加尖锐、狭窄,表明(002)晶面的晶面间距d(002)减小,碳材料经热处理后有序化程度和石墨化程度增加[23].根据Maire公式[24],XC⁃72 CB经1700℃处理后d(002)约为0.345 nm,与TEM表征结果一致.
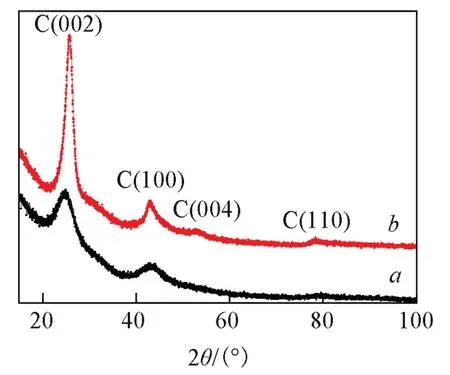
Fig.2 XRD patterns of XC⁃72 CB(a)and GCB(b)

Fig.3 Raman spectra of XC⁃72 CB(a)and GCB(b)
2.1.3 拉曼光谱表征 图3是XC⁃72 CB在1700℃热处理前后的拉曼光谱图.1700℃热处理后XC⁃72CB后,在2700 cm-1左右出现了明显的2D峰.GCB在1333和1584 cm-1左右出现的D峰和G峰与XC⁃72 CB相比变得更狭窄且尖锐.通过拟合得出GCB的G峰、D峰和2D峰的半峰宽都明显减小(表1).而半峰宽越小,说明碳材料的石墨化程度越高.此外,衍射峰强度比ID/IG的减小及I2D/IG的增加都说明了热处理后XC⁃72 CB的无序化程度降低,石墨化程度提高,这与XRD和TEM的结论一致,即热处理后XC⁃72 CB的石墨化程度明显提高.

Table 1 Raman spectroscopy data of XC⁃72 CB before and after 1700℃treatment
2.1.4 孔径及比表面积分析 高温热处理后,XC⁃72 CB的比表面积从235 m2/g减小至93 m2/g,最可几孔径从1.91 nm增大至2.71 nm,从微孔增大至介孔,这也充分说明微孔的减少导致GCB的比表面积减小[25].虽然微孔坍塌可能导致催化剂材料如Pt的负载减少,但由于反应介质很难进入微孔结构内部,导致负载于微孔之上的催化剂很难参与反应,故Pt纳米粒子只是需要合理的微孔数量和孔径分布,而不是越多越好.
2.1.5 热稳定性分析 图4(A)是XC⁃72 CB和GCB的热重(TG)分析曲线.可见,升温到570℃左右时,XC⁃72 CB的分解速率最大,质量发生明显减少;而GCB质量的显著变化开始于700℃左右,表明经1700℃的热处理后,GCB的热稳定性提高.两者的分解速率都是先慢后快,直至质量降为0.图4(B)是XC⁃72 CB热处理前后的差示扫描量热(DSC)曲线.XC⁃72 CB在576.7和796.4℃出现明显的峰;而GCB 2个峰都明显后移,出现在646.6和826.4℃,表明GCB的热稳定性提高.
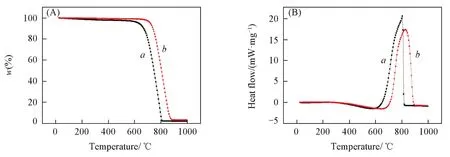
Fig.4 TG(A)and DSC(B)curves of XC⁃72 CB(a)and GCB(b)
2.2 载体预处理对催化剂性能的影响
2.2.1 预处理后GCB的结构表征 图5是GCB在不同条件下预处理后的红外(IR)光谱图.红外光谱的振动分成4个区域:600~1500 cm-1是C—C,C—O,C—N等的伸缩振动区域;1500~2000 cm-1是双键的伸缩振动区域,羟基和羧基的振动都在此区域;2000~2500 cm-1是累积双键和三键的伸缩振动区域;2500~4000 cm-1是X—H单键的伸缩振动区域.由图5可见,4个样品在3600和2900 cm-1处分别都有游离态的O—H和C—H的伸缩振动峰.经HNO3,H2SO4和H2SO4+HNO3预处理后,在1698~1735 cm-1区间出现羧基和羟基的伸缩振动峰,且混酸处理后峰强度最大.说明预处理试剂的不同,羟基与羧基的强度存在差异.

Fig.5 IR spectra of GCB before and after pretreatmenta.GCB;b.HNO3⁃GCB;c.H2SO4⁃GCB;d.OGCB.
图6是GCB经不同条件预处理后的拉曼光谱图.经HNO3,H2SO4和H2SO4+HNO3处理后,随着GCB表面的氧化,从2500 cm-1以后基线受含氧官能团的影响而上扬.D峰,G峰和2D峰的拉曼位移没有变化,证明预处理条件没有明显破坏碳材料的有序化程度.
2.2.2 催化剂的表征 图7是经过不同酸处理后的载体负载Pt催化剂后的TEM照片.由图7可见,4种催化剂中的Pt纳米颗粒分布较均匀,没有明显的集聚现象.Pt/GCB,Pt/HNO3⁃GCB,Pt/H2SO4⁃ GCB和Pt/OGCB中Pt的平均粒径分别为2.66,2.45,2.29,2.28 nm(图8),其中Pt/OGCB中Pt的颗粒尺寸最小,Pt的利用率最高.经酸处理后,GCB表面带上羟基和羧基官能团,增加了碳表面的负电荷量,GCB颗粒之间的静电斥力增强[26].
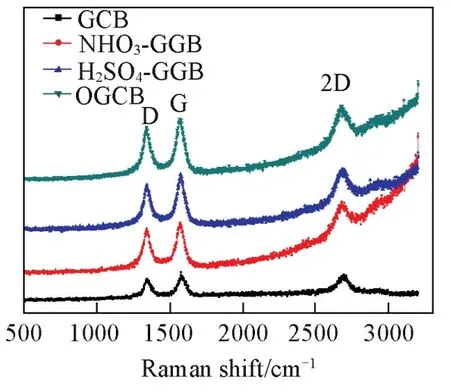
Fig.6 Raman spectra of GCB before and after pretreatmenta.GCB;b.HNO3⁃GCB;c.H2SO4⁃GCB;d.OGCB.

Fig.7 TEM images of different catalysts(A)Pt/GCB;(B)Pt/HNO3⁃GCB;(C)Pt/H2SO4⁃GCB;(D)Pt/OGCB.

Fig.8 Particle size distribution of different catalysts(A)Pt/GCB;(B)Pt/HNO3⁃GCB;(C)Pt/H2SO4⁃GCB;(D)Pt/OGCB.
由Pt/GCB,Pt/HNO3⁃GCB,Pt/H2SO4⁃GCB和Pt/OGCB 4种催化剂的X射线光电子能谱(XPS)全谱图[图9(A)]均可观察到Pt4f,C1s和O1s的光电子峰.4种催化剂中Pt4f的摩尔分数分别是0.59%,0.50%,0.50%和0.60%;O的摩尔分数分别是20.18%,17.34%,19.70%和19.55%.未经酸处理的GCB含有很多在制备过程中生成的不稳定的氧化物杂质,酸处理后GCB上的杂质被去除,同时引入了新的含氧基团(如羧基和羟基),导致催化剂中氧的含量发生变化.图9(B)是Pt/OGCB的C1s分谱图,结合能284.8和288.5 eV处分别对应C—C和基团,其中sp2杂化C所占比例为85.6%,说明C原子主要以π键的形式结合在一起,有序化程度较高.混合酸处理后,GCB表面杂质被基本去除,同时引入的基团能够提高载体在胶体中的稳定性,从而提高Pt纳米颗粒的稳定性[27].图9(C)~(F)分别是Pt/GCB,Pt/HNO3⁃GCB,Pt/H2SO4⁃GCB和Pt/OGCB催化剂的Pt4f分谱图.Pt(0)所占比例分别为77.67%,82.38%,86.77%和92.84%,说明酸处理后,Pt(0)所占比例增加,且使用不同的酸处理载体后合成的催化剂中Pt(0)的含量存在差异.

Fig.9 Full scan XPS spectra of the four catalysts(A),C1sXPS spectra of Pt/OGCB catalyst(B)and Pt4fXPS spectra of Pt/GCB(C),Pt/HNO3⁃GCB(D),Pt/H2SO4⁃GCB(E)and Pt/OGCB(F)catalyst(A)a.Pt/GCB;b.Pt/HNO3⁃GCB;c.Pt/H2SO4⁃GCB;d.Pt/OGCB.
2.2.3 催化剂的电化学性质 图10是不同酸处理的GCB负载Pt后所得催化剂的循环伏安曲线.可见,在饱和N2气氛围下,0.1 mol/L高氯酸电解质溶液中,CV曲线氢的吸附⁃脱附峰均出现在0~0.3 V之间.混合酸可生成较多的和官能团,Pt(0)比例增加,氢吸附电势转移到较低电势,氧的氧化还原峰出现在0.8 V左右.Pt/GCB,Pt/HNO3⁃GCB,Pt/H2SO4⁃GCB和Pt/OGCB的ECSA分别为38.10,48.94,51.22和61.69 m2/g,可见,负载Pt后所得基催化剂的ECSA根据不同处理条件有不同程度的增加.混合酸处理后合成的催化剂表现出最大的电化学活性表面积.酸处理GCB后增加了其负载Pt颗粒的活性位点,而这些活性位点是含氧官能团或载体表面的缺陷位点.
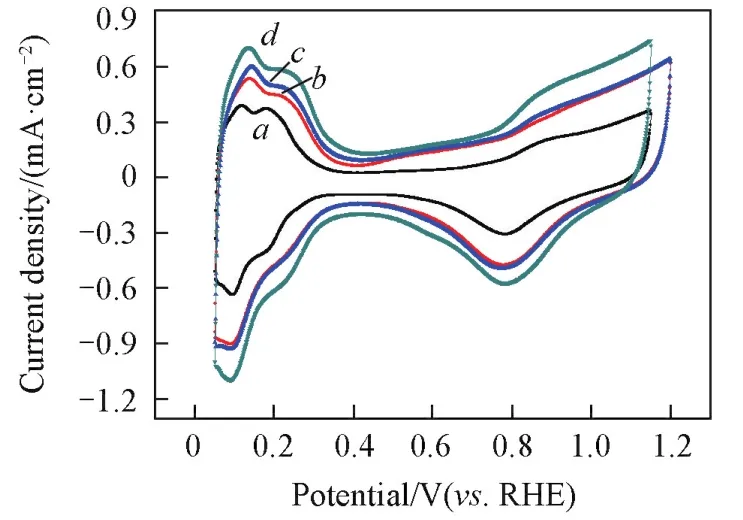
Fig.10 CV curves of catalysts Pt/GCB(a),Pt/HNO3⁃GCB(b),Pt/H2SO4⁃GCB(c)and Pt/OGCB(d)
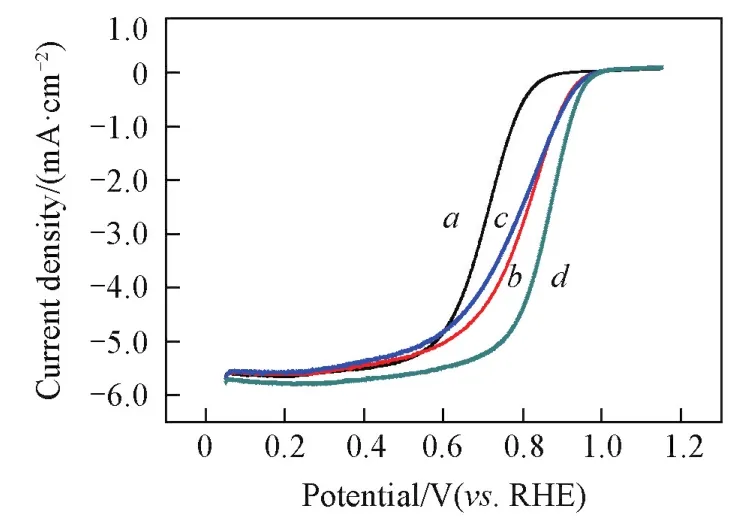
Fig.11 LSV curves of catalysts Pt/GCB(a),Pt/HNO3⁃GCB(b),Pt/H2SO4⁃GCB(c)and Pt/OGCB(d)
图11是载体经不同酸处理后负载Pt所得催化剂的LSV图.可见,ORR起始电位均为1.0 V;极限电流密度均在5.5~6.0 mA/cm2之间.0.9 V下,Pt/GCB,Pt/HNO3⁃GCB,Pt/H2SO4⁃GCB和Pt/OGCB的电流密度分别是0.15,0.64,0.66和1.48 mA/cm2,质量比活性依次是0.010,0.040,0.041和0.093 A/mg.载体预处理后催化剂的ORR活性明显增加,且在预处理试剂为硫酸和硝酸的混合溶液时,制备的催化剂质量比活性最大.混合酸预处理GCB后生成和比例增加,带入的含氧基团能促进修饰后的GCB表面和锚定的催化剂金属离子之间发生化学反应,形成成核前驱体:

式中,M+为金属离子,X-为阴离子.同时伴随着较低的阻抗和迅速的电子传导.嫁接羟基和羰基后,附着在GCB表面的Pt离子相比于C更易被还原[28].
2.3 Pt/OGCB与商业催化剂的性能对比
图7(D)和图12是Pt/OGCB催化剂与商业Pt/C(JM)催化剂经5000周加速老化前后的TEM照片.可见,经5000周加速老化后,催化剂颗粒出现了少量团聚.Pt/OGCB催化剂的平均粒径由2.28 nm[图8(D)]增大至3.67 nm[图13(A)],增大了54%;商业催化剂Pt/C(JM)的平均粒径由2.50 nm增大至3.97 nm[图13(B,C)],增大了59%.加速老化后,自制催化剂和商业催化剂中Pt颗粒的平均粒径都增加,但商业催化剂中Pt的长大或积聚程度高于自制Pt/OGCB催化剂,可见自制Pt/OGCB催化剂的Pt颗粒增长速度低于商业催化剂.

Fig.12 TEM images of Pt/OGCB catalysts and Pt/C(JM)after 5000 cycles accelerated durability tests(A)Pt/OGCB(after 5000 cycles);(B)Pt/C(JM)(fresh);(C)Pt/C(JM)(after 5000 cycles).

Fig.13 Prticle size distribution of Pt/OGCB catalysts and Pt/C(JM)after 5000 cycles accelerated durability tests(A)Pt/OGCB(after 5000 cycles);(B)Pt/C(JM)(fresh);(C)Pt/C(JM)(after 5000 cycles).
图14是商业Pt/C(JM)催化剂和自制Pt/OGCB催化剂经电化学加速老化5000周前后的CV曲线.可见,在N2气饱和氛围的高氯酸电解质中,2种催化剂的氢吸附⁃脱附峰都出现在0~0.3 V之间,且都有明显的双峰形状;Pt/OGCB氧的还原峰位于0.8 V左右,与商业催化剂Pt/C(JM)类似.5000周加速老化前后,Pt/OGCB催化剂的ECSA分别为75.25和62.26 m2/g,衰减了17.3%;Pt/C(JM)催化剂的ECSA分别是56.91和42.62 m2/g,衰减了25.1%.可见,Pt/OGCB的ECSA衰减程度比Pt/C(JM)低,这可能是因为石墨化后,对液相传质不利的微孔结构减少,使负载于碳材料表面的Pt能与反应物充分接触;另一方面,载体石墨化后,耐久性明显增加.因此,Pt/OGCB催化剂的电化学活性和电化学稳定性都优于商业Pt/C(JM)催化剂.
图15是商业Pt/C(JM)催化剂和Pt/OGCB催化剂电化学加速老化5000周前后的LSV曲线.可见,2种催化剂初始极限电流密度均在5.0~6.0 mA/cm2之间,氧化⁃还原反应(ORR)起始电位均为1.0 V.通过计算0.9 V下催化剂的ORR性能可知,Pt/OGCB催化剂和商业Pt/C(JM)催化剂的质量比活性(MA)分别是0.093和0.073 A/mg,5000周加速老化后,MA降至0.065和0.042 A/mg,分别衰减了29.5%和42.5%,Pt/OGCB催化剂表现出优于商业Pt/C(JM)催化剂的ORR活性和ORR稳定性.

Fig.14 CV curves of catalysts befor(a)and after(b)5000 cycles accelerated durability tests(A)Pt/OGCB;(B)Pt/C(JM).

Fig.15 LSV curves of catalysts before(a)and after(b)5000 cycles accelerated durability tests(A)Pt/OGCB;(B)Pt/C(JM).
2.4 Pt/OGCB催化剂在燃料电池中的应用
图16(A)和(B)分别为50 mA/cm2负载下MEAPt/C(JM)和MEAPt/OGCB单电池0,300,1200和1500次启停循环后的极化曲线和功率密度曲线.每次循环的具体操作为:PEMFC开机后通气,在开路电压(OCV)状态下保持25 s,关闭气体的同时打开辅助负载(恒电流模式2.5 A,即50 mA/cm2),直至电压下降至0.1 V以下关闭辅助负载,再通入气体进入下一循环.考虑2张膜都要加载至耗尽气体且控制变量的前提,设置2张膜的单次循环时间均为50 s.启停循环实验共进行1500次循环,合计20.83 h.
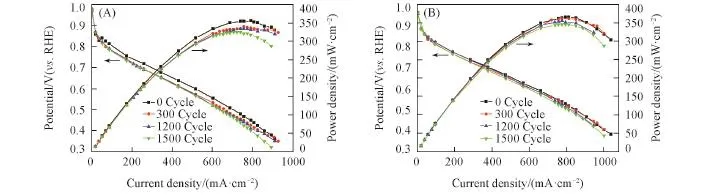
Fig.16 Single⁃cell performance of MEAPt/C(JM)(A)and MEAPt/OGCB(B)
由图16可见,随着循环次数的增加,MEAPt/C(JM)单电池性能有不同程度的衰减.MEAPt/OGCB的最大功率密度衰减比MEAPt/C(JM)的小,说明MEAPt/OGCB耐久性较好.在低电流密度0.4 A/cm2下,前300次启停循环中MEAPt/C(JM)的电势衰减了3.4%,即0.077 mV/cycle;经过1500次启停循环后总的衰减率也只有3.8%,即0.017 mV/cycle.MEAPt/OGCB在0~300次循环中只衰减了1.7%,即0.037 mV/cycle,低于商业催化剂的衰减率,经过1500次启停循环后,衰减了3.0%,即0.013 mV/cycle,与商业催化剂大体相同,随着启停次数的增加衰减率逐渐降低.可见,在低电流密度下,MEAPt/OGCB的耐久性略高于MEAPt/C(JM).在高电流密度0.9 A/cm2下,MEAPt/C(JM)的最大功率密度在0~300次循环后衰减了4.4%,即0.07 mV/cycle;1500次启停循环后总的衰减率是9.2%,即0.03 mV/cycle;而MEAPt/OGCB经历1500次启停循环后总的衰减率是6.1%,即0.017 mV/cycle.可见,在高电流密度下,Pt/OGCB催化剂的耐久性明显优于商业催化剂Pt/C(JM).
3 结 论
为了提高GCB负载Pt的能力,用不同的酸处理GCB.结果表明,使用浓硫酸和浓硝酸的混合酸处理GCB后,制得的Pt/OGCB催化剂电化学性能最佳,ECSA和MA均优于商业Pt/C(JM)催化剂,且5000周加速CV老化后,Pt/OGCB的ECSA和MA只分别衰减了17.3%和29.5%,低于Pt/C(JM)的25.1%和42.5%.将优化后的Pt/OGCB催化剂和商业Pt/C(JM)催化剂用作燃料电池的阴极,应用于单电池,经1500次启停循环后,在高电流密度下,MEAPt/OGCB在相同电流密度下电势的衰减率(6.1%)低于MEAPt/C(JM)(9.2%),且在低电流密度下得到相似的结果.与商业Pt/C(JM)催化剂相比,Pt/OGCB催化剂在保证较高电化学性能的前提下提高了催化剂的耐久性,更能适应PEMFC复杂的工况环境.
[1] Chen K.C.,Ji M.D.,Chem.J.Chinese Universities,2016,37(5),989—995(陈康成,纪梦蝶.高等学校化学学报,2016,37(5),989—995)
[2] Huang Z.,Lin R.,Tang W.C.,Ma J.X.,Chin.J.Power Sources,2014,38(1),174—177(黄真,林瑞,唐文超,马建新.电源技术,2014,38(1),174—177)
[3] Smimova A.,Dong X.,Hara H.,Vasiliev A.,Sammes N.,Int.J.Hydrogen Energy,2005,30(2),149—159
[4] Kim J.,Lee J.,Tak Y.,J.Power Sources,2009,192(2),674—678
[5] Sasaki K.,Shao M.,Adzic R.,Dissolution and Stabilization of Platinum in Oxygen Cathodes,Springer,New York,2009,7—27
[6] Bruijn F.A.D.,Dam V.A.T.,Janssen G.J.M.,Fuel Cells,2008,8(1),3—22
[7] Zhang L.,Lin R.,Huang Z.,Fan R.J.,J.Fuel Chem.Technol.,2015,(3),352—359(张路,林瑞,黄真,范仁杰.燃料化学学报,2015,(3),352—359)
[8] Lei M.,Liang C.,Wang Y.,Huang K.,Ye C.,Liu G.,Wang W.,Jin S.,Zhang R.,Fan D.,Electrochim.Acta,2013,113,366—372
[9] Zhang Y.,Pan Y.,Liu J.,Wang G.L.,Cao D.X.,Chem.Res.Chinese Universities,2015,31(1),117—122
[10] Su F.,Zeng J.,Bao X.,Yu Y.,And J.Y.L.,Zhao X.S.,Chem.Mater.,2005,117(15),3960—3967
[11] Yang M.N.,Lin R.,Fan R.J.,Zhao T.T.,Zeng H.,Acta Phy⁃Chim.Sin.,2015,31,2131—2138(杨美妮,林瑞,范仁杰,赵天天,曾浩.物理化学学报,2015,31(11),2131—2138)
[12] Wang C.G.,Pan X.,Zhang L.,Zhu M.K.,Li D.K.,Diao L.B.,Li W.Y.,Chem.J.Chinese Universities,2015,36(2),368—374(王存国,潘璇,张雷,朱孟康,李德凯,刁玲博,李伟彦.高等学校化学学报,2015,36(2),368—374)
[13] Sevilla M.,Fuertes A.B.,Carbon,2006,44(3),468—474
[14] Wang X.,Hsing I.M.,Yue P.L.,J.Power Sources,2001,96(2),282—287
[15] Meyers J.P.,Darling R.M.,J.Electrochem.Soc.,2005,153(8),A1432—A1442
[16] Meng Q.,Bai J.,Guo S.,Chunping L.I.,Chem.Res.Chinese Universities,2015,31(6),1072—1077
[17] Zhao X.,Hayashi A.,Noda Z.,Kimijima K.I.,Yagi I.,Sasaki K.,Electrochim.Acta,2013,97(5),33—41
[18] Mench M.M.,Kumbur E.C.,Veziroglu T.N.,Polymer Electrolyte Fuel Cell Degradation,Academic Press,New York,2011,443—460
[19] Yano H.,Kataoka M.,Yamashita H.,Uchida H.,Watanabe M.,Langmuir,2007,23(11),6438—6445
[20] Hara M.,Lee M.,Liu C.H.,Chen B.H.,Yamashita Y.,Uchida M.,Uchida H.,Watanabe M.,Electrochim.Acta,2012,70(6),171—181
[21] Yano H.,Akiyama T.,Bele P.,Uchida H.,Watanabe M.,Phys.Chem.Chem.Phys.,2010,12(15),3806—3814
[22] Kim J.Y.,Lee S.,Kim T.Y.,Kim H.T.,Electrochim.Acta,2014,134(21),418—425
[23] Liang C.,Dai S.,Guiochon G.,Anal.Chem.,2003,75(18),4904—4912
[24] Bragg W.L.,Proc.Carnbridge Philos.Soc.,1913,17,43—57
[25] Saito T.,Matsushige K.,Tanaka K.,Phys.Rev.B:Condens.Matter,2002,323(1),280—283
[26] Chiang Y.C.,Ciou J.R.,Int.J.Hydrogen Energy,2011,36(11),6826—6831
[27] Kim T.W.,Park I.S.,Ryoo R.,Angew.Chem.,2003,115(36),4511—4515
[28] Li L.X.,Feng L.,New Carbon Mater.,2011,26(3),224—228
(Ed.:S,Z,M)
†Supported by the National Natural Science Foundation of China(No.21276199),the Fundamental Research Funds for the Central Universi⁃ties,China(No.0500219216),the Young Talents“Climbing”Program of Tongji University and the Programme of Introducing Talents of Discipline to Universities,China(No.B08019).
Effect of Acid⁃treatment of Graphitized Carbon Supports on Performance of Fuel Cell Catalysts†
YAN Haixu1,2,YANG Meini1,2,ZENG Hao1,2,PU Hongting3,LIN Rui1,2∗
(1.Clean Energy Automotive Engineering Center,Tongji University,Shanghai 201804,China;2.School of Automotive Studies,Tongji University,Shanghai 201804,China;3.School of Materials Science and Engineering,Tongji University,Shanghai 201804,China)
The graphitized carbon black(GCB)was obtained by a high temperature(1700℃)treatment of the XC⁃72 commercial carbon black(XC⁃72 CB).The functional groups of GCB were modified by acid treat⁃ment.Transmission electron microscopy(TEM),X⁃ray diffraction,Raman spectrum and infrared spectroscopy displayed the GCB with acid treatment had a higher degree of graphitization.Oxygen⁃containing functional groups were introduced into the GCB surface and the ordered structure of the GCB was maintained at the same time.Nitrogen adsorption and desorption experiment showed the GCB had smaller specific surface area and less micropore compared to XC-72 CB.Thermogravimetric analysis showed that GCB had the better heat stability. Cyclic voltammetry and linear sweep voltammetry test showed that the electrochemical specific activity area(ECSA)(75.25 m2/g)and the mass activity(MA)(0.093 A/mg)of conc.H2SO4and conc.HNO3treated GCB(abbreviated as OGCB)were higher than those of the commercial one.TEM showed the Pt/OGCB had an average particle diameter of 2.28 nm,smaller than the commercial one.After durability test of 5000 cycles,the ECSA and MA of Pt/OGCB decreased by 17.3%and 29.5%,respectively,smaller than those of Pt/C(JM)(25.1%and 42.5%).Both activity and durability performance of Pt/OGCB catalyst were better than those of commercial Pt/C(JM)catalyst in oxygen reduction reaction.In addition,in single cell test,the durability of Pt/OGCB catalyst was also better than that of the commercial catalyst.The results show that OGCB has a promising application prospect in the field of proton exchange membrane fuel cell(PEMFC)catalyst support.
Proton exchange membrane fuel cell;Graphitized carbon black;Acid treatment;Durability
O646
A
10.7503/cjcu20160426
2016⁃06⁃13.网络出版日期:2016⁃11⁃22.
国家自然科学基金(批准号:21276199)、中央高校基本科研基金(批准号:0500219216)和同济大学青年英才计划攀登高层项目及高等学校学科创新引智计划项目(批准号:B08019)资助.
联系人简介:林 瑞,女,博士,教授,主要从事新能源技术方向新材料及燃料电池技术研究.E⁃mail:ruilin@tongji.edu.cn
