镉暴露致大鼠肾损伤的量效关系及其机制
2016-12-09王乐乐刘江正刘萌萌孔德钦于卫华张晓迪海春旭
王乐乐,刘江正,刘萌萌,孔德钦,张 涛,于卫华,刘 瑞,张晓迪,王 欣,海春旭*
(第四军医大学预防医学院毒理学教研室,陕西省自由基生物学与医学重点实验室,陕西 西安 710032)
镉暴露致大鼠肾损伤的量效关系及其机制
王乐乐,刘江正,刘萌萌,孔德钦,张 涛,于卫华,刘 瑞,张晓迪,王 欣,海春旭*
(第四军医大学预防医学院毒理学教研室,陕西省自由基生物学与医学重点实验室,陕西 西安 710032)
镉污染的危害已经成为全球普遍关注的环境问题。肾脏是镉中毒的靶器官,是镉毒性防治研究的重点[1-2]。研究进展表明,镉暴露产生的毒性与氧化损伤有关。而氧化应激或损伤主要与活性氧(reactive oxygen species,ROS)水平密切相关,活性氧既有内源性的也有外源性的,但均与线粒体的功能和结构具有内在联系。目前镉致肾线粒体损伤的机制还不太清楚,故本研究采用CdCl2对SD大鼠进行28 d染毒试验,旨在观察大鼠亚急性镉暴露致肾损伤的量效关系和毒理机制。
1 材料与方法
1.1 主要试剂与仪器
氯化镉(CdCl2·2.5H2O),分析纯,购自国药集团化学试剂有限公司(批号F20100208);尿素氮(urea)、肌酐(creatinine,苦味酸法)测定试剂盒购自北京利德曼生化股份有限公司;DHE、MitoSOX、蛋白酶抑制剂Cocktail和染核试剂Hoechst 33528购自美国Sigma公司;牛血清白蛋白购自美国Boehringer公司;BCA蛋白定量试剂盒购自Thermo公司;兔抗 SOD1、SOD2购于武汉华美生物工程有限公司;兔抗GPx-1抗体购于Bioworld公司;兔抗CAT抗体购于Protein Tech公司;兔抗Bcl-2购于Cell Signal Technology公司;兔抗Bax抗体购于Santa公司。
日立7020全自动生化检测仪购自日立公司;全波段酶标仪为TECAN公司产品;正置荧光显微镜、激光共聚焦显微镜为Olympus公司产品。透射电镜观察在口腔医院重点实验室进行。Mini-Protean电泳系统、蛋白半干转装置及凝胶成像系统均为Bio-Rad公司产品。
1.2 实验方法
1.2.1 动物与饲养 SPF级雄性SD大鼠40只,体质量100~150 g,由第四军医大学实验动物中心提供。饲养于SPF级动物房,自由摄食、饮水。实验动物生产许可证号为SCXK(陕)2014-0490,动物房温度为(24±2)℃,相对湿度为50%±10%。
1.2.2 动物分组和处理 大鼠适应性喂养5 d后,随机分为4组:对照组(去离子水);低剂量镉(1mg/kg CdCl2)处理组;中剂量镉(3mg/kg CdCl2)处理组;高剂量镉(5mg/kg CdCl2)处理组。灌胃体积为10mL/kg。灌胃顺序按照对照组、低、中、高剂量顺序依次进行,给药剂量根据文献并结合课题组前期工作和预实验确定。每天灌胃1次,连续染毒4周后,禁食12h,腹腔注射2%戊巴比妥钠,麻醉后腹主动脉取血。用经高温处理过的器械剪取双侧肾脏,称湿质量,计算肾脏系数(双侧肾脏湿质量/大鼠体质量)。取左肾分为3部分(主要为肾皮质),分别制作冰冻切片、石蜡切片(4%甲醛固定)和透射电镜标本(2%戊二醛固定)。右肾于-80℃保存备用。
1.2.3 血清尿素氮(BUN)和肌酐(SCr)含量检测 未经抗凝的全血经3 500 r/min离心10min后,取上部血清,用日立7020全自动生化仪测定血清中BUN和SCr含量。
1.2.4 HE染色[3]肾组织用4%甲醛溶液固定,常规脱水、包埋、切片、脱蜡、HE染色、脱水、封片后于正置荧光显微镜下观察并拍照。
1.2.5 透射电镜观察 肾皮质切成1mm3左右的立方体,置于2%戊二醛中固定,制作肾脏电镜切片,透射电镜下观察各组大鼠肾近曲小管上皮细胞的超微病理变化。
1.2.6 肾皮质ROS水平检测 采用荧光探针DHE检测细胞内ROS水平[4],采用MitoSOX用于检测线粒体内ROS水平[5],具体方法如下:制备厚度为10μm的肾组织冰冻切片,置于湿盒中,将10μmol/L DhE、5mmol/LmitoSOX分别按1∶1 000、1∶500的比例用PBS稀释,并将10μmol/L的Hoechst按1∶500的比例加入稀释过的两种染料中混匀,向每张切片上的肾组织滴加混合液150μL,在37℃孵箱中避光孵育40min,PBS洗3次,甘油封片,于激光共聚焦显微镜下观察结果并拍照。
1.2.7 肾组织SOD1、SOD2、CAT、GPx-1和GRP78、Bax、Bcl-2蛋白表达的测定 用眼科剪将100mg肾皮质部分剪碎,置于玻璃匀浆器中,加入1mL裂解缓冲液(1mL RIPA裂解液+20μL矾酸钠+5μL Cocktail),冰上充分匀浆,4℃裂解30min,匀浆液于4℃、10 000 r/min离心20min,取上清,留取20μL用
作BCA蛋白定量,余下部分加入等体积2×SDS和5μL二硫苏糖醇(dithiothreitol,DTT),100℃煮沸5min,分装后保存于-80℃ ,用于常规Western blotting测定。
1.3 统计学方法
2 结果
2.1 一般情况观察
对照组大鼠摄食、摄水情况均正常,镉暴露组大鼠随CdCl2剂量升高逐渐出现食欲减退、皮毛粗糙、情绪暴躁等现象。染毒期间各组大鼠体质量随时间的变化如图1所示。与对照组比较,各镉暴露组大鼠体质量及肾脏脏器系数(表1)的差异均无统计学意义(P均>0.05)。
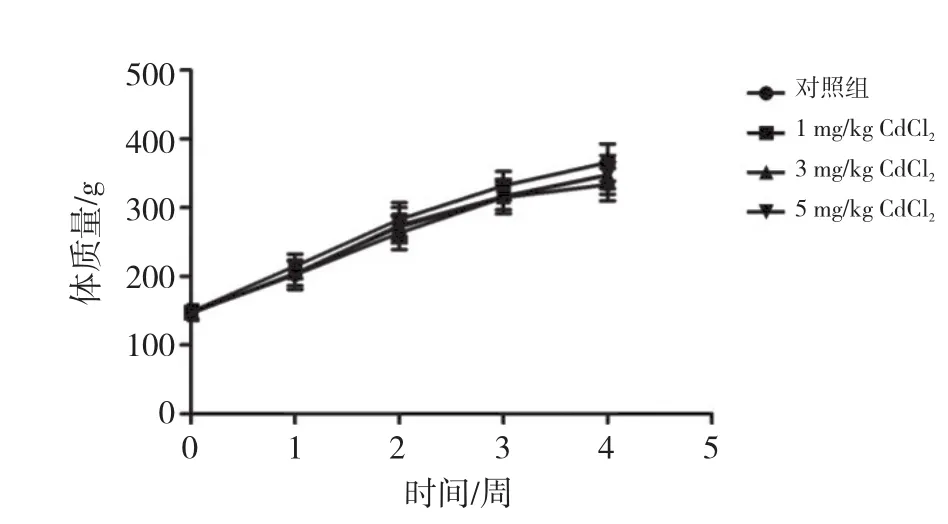
图1 大鼠体质量增长曲线
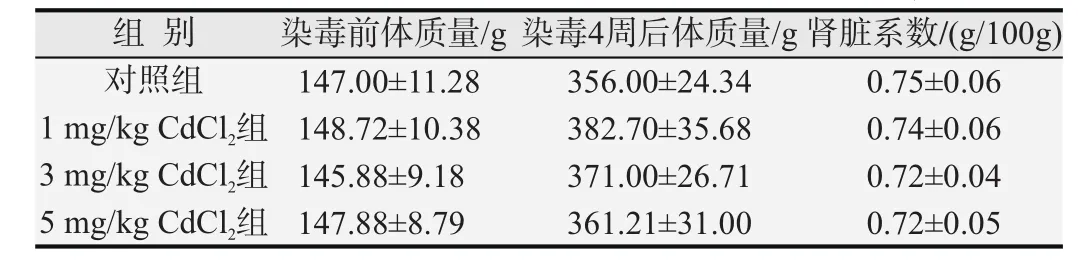
表1 各组大鼠体质量变化和造模结束时的肾脏脏器系数(n=10)
2.2 肾功能变化
通过测定血清中BUN和SCr的含量评价大鼠肾功能[6]。CdCl灌胃4周后,血清中BUN含量随镉暴露剂量升高逐渐增加(r=0.463)。其中,5mg/kg CdCl2暴露组与对照组相比,BUN含量显著升高(P<0.05),提示出现肾功能损伤。但各剂量组SCr含量与对照组相比差异均无统计学意义(P>0.05)。见表2。

表2 镉暴露对大鼠肾功能的影响(n=10)
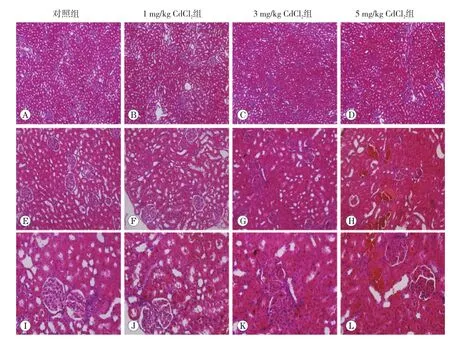
图2 肾脏病理变化(HE染色)
2.3 肾组织病理学观察
HE染色见图2。对照组大鼠肾脏组织结构完整,肾小球形状规则,毛细血管网轮廓清晰。肾小管轮廓
清楚,游离面刷状缘清晰,上皮细胞排列整齐,管腔干净。随着CdCl2染毒剂量增加,肾脏病理损伤进行性加重。1mg/kg CdCl2处理组肾小球结构尚完整,肾小管略肿胀,间质充血。3mg/kg CdCl2处理组肾小球肿胀,结构破坏严重,细胞数目增多,排列紊乱,球囊间隙几乎不见;肾小管明显肿胀,管腔狭窄,上皮细胞坏死脱落堆积于管腔;肾间质充血,炎细胞浸润。5mg/kg CdCl2处理组肾小球肿胀,细胞数目增多,毛细血管网出现裂隙;肾小管广泛性病变,排列紊乱、肿胀、管腔狭窄几乎不见,上皮细胞坏死、结构消失,脱落细胞堆积于管腔,伊红染色阳性;肾间质充血严重,炎性细胞浸润程度明显增加。
2.4 肾皮质超微结构的变化
超微结构观察结果见图3。镉暴露主要引起肾小管上皮细胞损伤,尤其是线粒体损伤严重,出现肿胀、变形、空泡样变,线粒体嵴紊乱、模糊甚至消失,损伤程度随CdCl2染毒剂量的增加而逐渐加重。
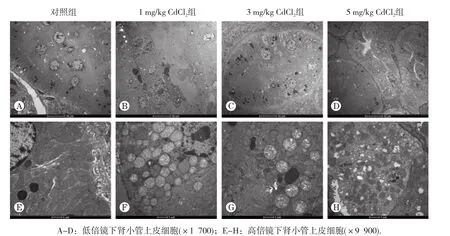
图3 肾皮质超微结构观察
2.5 肾组织细胞内ROS水平的变化
DHE和MitoSOX染色结果如图4所示,随CdCl2染毒剂量的增加,细胞内ROS水平逐渐增加,线粒体内ROS水平增加更为明显。

图4 肾脏冰冻切片
2.6 肾组织中SOD1、SOD2、CAT和GPX1蛋白的表达
如图5所示。与对照组比较,镉暴露组随CdCl2剂量升高,存在于线粒体嵴的抗氧化酶SOD2(r=0.854)与存在于细胞质中的CAT(r=0.975)和GPX1(r=0.918)蛋白表达均明显上调(P均<0.05);其中,1mg/kg CdCl2处理组GPX1蛋白表达量增加明显(P<0.05);3mg/kg CdCl2处理组CAT蛋白和GPX1蛋白表达量增加(P<0.05);5
mg/kg处理组SOD2、CAT、GPX1蛋白表达量均明显增加(P<0.05)。而存在于细胞质中的SOD1表达则逐渐下调(r=-0.987,P<0.05)。
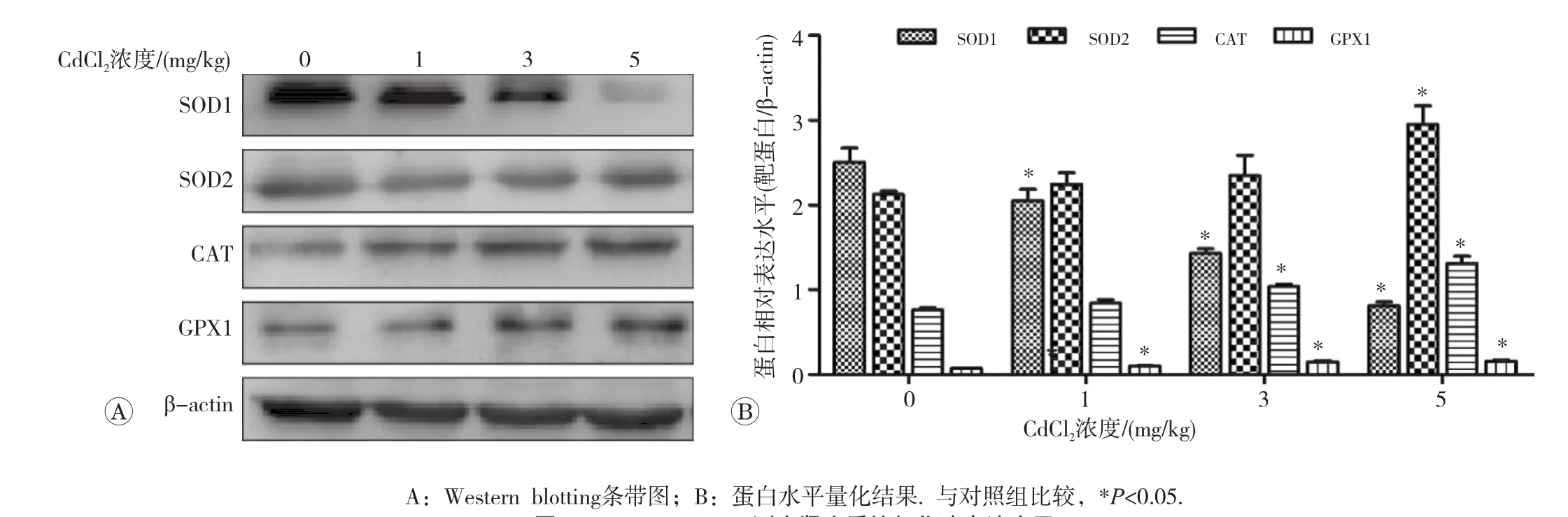
图5 Western blotting测定肾皮质抗氧化酶表达水平
2.7 肾组织中GRP78、Bax和Bcl-2蛋白的表达
通过Western blotting检测各剂量组肾皮质内质网应激标志蛋白GRP78的表达水平[7],促凋亡蛋白Bax和抑凋亡蛋白Bcl-2的表达水平。结果见图6。与对照组相比,GRP78表达水平随CdCl2剂量升高而增加(r= 0.913,P<0.05);1和3mg/kg CdCl2处理组Bax表达显著增加(r=0.226,P<0.05);Bcl-2表达随CdCl2剂量增加逐渐降低(r=-0.85,P<0.05),但Bax/Bcl-2随剂量增加而增加(r=0.83,P<0.05),见图7。
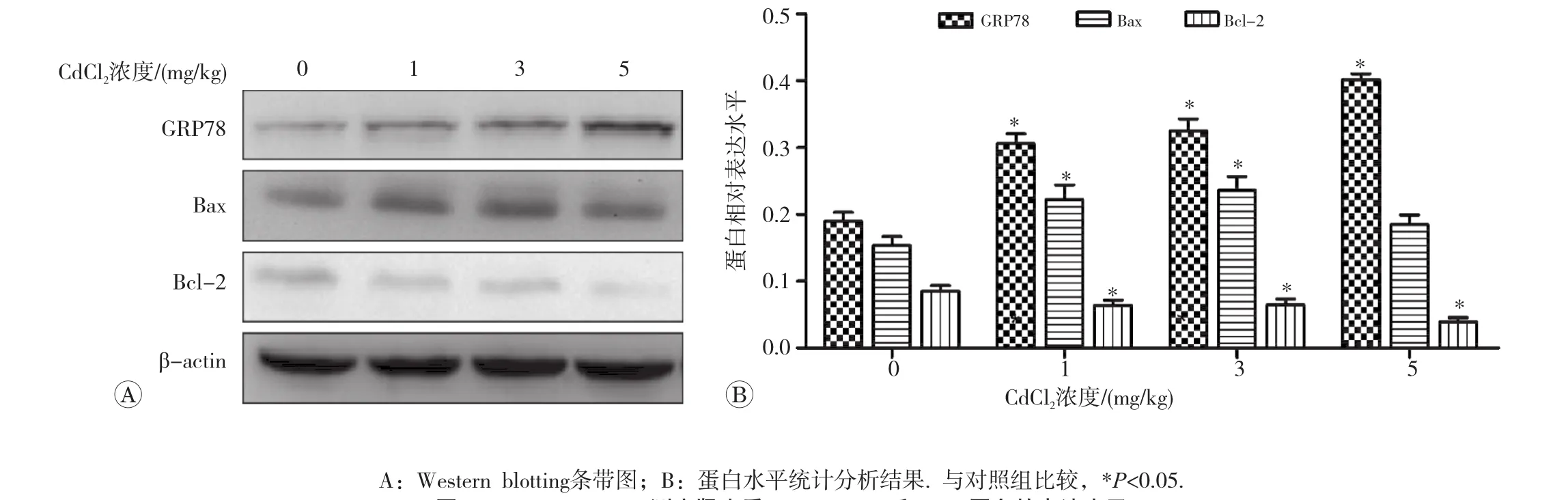
图6 Western blotting测定肾皮质GRP78、Bax和Bcl-2蛋白的表达水平
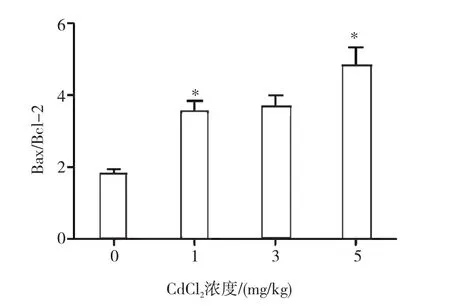
图7 Bax/Bcl-2值的变化
3 讨 论
人体非必需重金属元素镉(Cd),作为已确定的人类致癌物,容易进入空气、水源、土壤、动植物等造成环境污染,并经呼吸道、消化道、皮肤等途径间接进入人体,导致CdCl(Cd2+)中毒的发生。Cd2+经呼吸道或消化道摄入后进入血循环,血浆中的游离Cd2+随血液循环被输送到全身各脏器的组织细胞中,从而对肝脏、肾脏、骨骼、生殖系统、心血管系统和神经系统[8-9]等造成不同程度的损害。有研究显示,肾近端小管S1段可能是镉沉积的主要靶点,体内约30%的镉沉积于肾小管区[10],也有研究认为,Cd2+导致的骨软化症
等严重后果是继发于肾小管病的[11],这些使肾脏成为Cd2+对人体毒性作用的主要靶器官和研究热点。
本研究选取肾脏作为研究重点,经CdCl2灌胃染毒,并通过HE染色和检测大鼠血清肾功能指标BUN和SCr的结果,证明镉暴露可以造成大鼠肾脏结构和功能的损伤。且随着CdCl2染毒剂量的增大,肾脏病理损伤进行性加重。证明了镉暴露导致的肾脏损伤具有剂量依赖性。
同时,通过透射电镜观察发现,肾小管上皮细胞线粒体出现肿胀变圆、嵴消失、空泡样变等损伤,且随着染毒剂量增大,损伤程度也加重,提示Cd2+造成的大鼠肾毒性的关键靶点是线粒体。
线粒体作为生物体细胞的重要组成部分,其承担的呼吸作用是为机体提供ATP和维持各项生命活动所必需的[12],线粒体的结构损伤和功能障碍会导致机体的损伤,从而引发脏器损伤甚至全身疾病。
有研究证明Cd2+只有在进入细胞后才产生毒性[13],细胞凋亡是Cd2+细胞毒性的主要事件。因此本研究还测定了肾组织凋亡相关蛋白Bax和Bcl-2的表达水平,发现抑凋亡蛋白Bcl-2表达随CdCl2剂量增加逐渐降低(r=-0.85,P<0.05),1和3mg/kg CdCl2处理组促凋亡蛋白Bax表达逐渐增加(r=0.226,P<0.05),5mg/kg CdCl2处理组表达增高,但较前组表达低(P>0.05)。通常采用Bax/Bcl-2的值来观察细胞凋亡情况,实验结果发现虽然Bax在5mg/kg CdCl2处理组表达较前减少,但Bax/ Bcl-2 依然随CdCl2染毒剂量的增大而增加,提示肾组织细胞的凋亡程度可能存在剂量依赖性地增加。
目前关于Cd2+毒性的具体机制尚不清楚[14],但大量研究发现,氧化应激在镉暴露导致的肾损伤中起着重要作用[15]。氧化应激损伤主要表现为生物大分子(如DNA、蛋白质和脂类)的结构及功能的损伤。尽管Cd2+只有一个氧化态,不能直接产生自由基,但是Cd2+可通过从金属依赖性抗氧化酶(如SOD)中置换金属,干扰这些酶类的作用[16],也可结合细胞内含巯基的物质(如谷胱甘肽GSH、金属硫蛋白MT等)[14],间接促进ROS的产生,使机体氧化还原平衡被破坏[17],可导致线粒体功能障碍[18],抑制呼吸作用,诱发氧化应激和脂质过氧化等[19],直至引起机体不可逆的损伤。
本研究通过DHE和MitoSOX染色以及Western blotting观察到,肾组织的ROS水平呈剂量依赖性地增加,而抗氧化酶类的表达呈现不同趋势。SOD作为清除自由基的主要酶类,存在于细胞质中的抗氧化酶SOD1表现出下调趋势,这证明Cd2+肾毒性可能引起氧化还原稳态失衡导致氧化应激,从而引起主要抗氧化酶SOD1表达下调。而存在于线粒体嵴的抗氧化酶SOD2蛋白表达随镉暴露剂量增加明显上调,提示,线粒体尽管已经出现形态和功能损伤,但可能还处在氧化应激损伤代偿性修复范围。此外,SOD主要清除超氧阴离子自由基,而CAT,GPX1主要清除过氧化氢(H2O2)等[17],,SOD1表达下调证明镉暴露后细胞清除的能力下降,可能是由于细胞内ROS主要是大量堆积,超过了氧化应激损伤代偿性修复范围,而H2O2等还未超过代偿范围,从而使抗氧化酶SOD1与CAT、GPX1的表达呈现不同趋势。但其确切机制还有待于进一步深入研究。
研究中同时发现,肾皮质内质网应激标志蛋白GRP78表达水平随镉暴露剂量增加而进行性上调,提示内质网应激在Cd2+中毒致肾损伤中可能发挥作用。
随着工业化进程的加快,Cd污染已成为严重危害人类健康的全球性环境问题,Cd导致的食品污染更是已成为严重公害,尤其是Cd2+中毒尚无有效的治疗方案[15]。本研究观察到亚急性镉暴露造成的大鼠肾损伤存在剂量依赖性,氧化应激包括线粒体和内质网应激可能是Cd2+中毒损伤的关键机制。这些发现对于寻找切实有效的治疗Cd2+中毒的药物和干预措施提供了思路,有着一定的现实意义。
[1] JohrIn,Jacquillet G,Unwin R.Heavymetal poisoning:the effects of cadmium on the kidney[J].Biometals,2010,23(5):783-792.
[2] Yangh,Shu Y.Cadmium transportersin the kidney and cadmium-induced nephrotoxicity[J].Int Jmol Sci,2015,16(1):1484-1494.
[3] Wang Y,Wu Y,Luo K,etal.The protective effects of selenium on cadmium-induced oxidative stress and apoptosis viamitochondria pathway inmice kidney[J].Food Chem Toxicol,2013,58:61-67.
[4] Wang C,Blough ER,Arvapalli R,etal.Metabolic syndrome-induced tubulointerstitial injury:Role of oxidative stress and preventive effects of acetaminophen[J].Free Radic Biolmed,2013,65:1417-1426.
[5]mitchell T,Sabah,Laakman J,etal.Role ofmitochondrialderived oxidantsin renal tubular cell cold-storage injury[J].Free Radic Biolmed,2010,49(8):1273-1282.
[6] Latchoumycandane C,Nagy LE,McIntyre TM.Chronic ethanol ingestion induces oxidative kidney injury through taurine-inhibitable inflammation[J].Free Radic Biolmed,2014,69:403-416.
[7] Xu W,Neckers L.Gr(i)p the ER to stress outmelanoma[J].
Cancer Cell,2016,29(6):769-771.
[8] Chen S,Ren Q,Zhang J,etal.N-acetyl-L-cysteine protects against cadmium-induced neuronal apoptosis by inhibiting ROS-dependentactivationof Akt/mTOR pathway inmouse brain[J].Neuropath Appl Neuro,2014,40(6):759-777.
[9] Zhai Q,Narbad A,Chen W.Dietary strategies for the treatmentof cadmium and lead toxicity[J].Nutrients,2015,7(1):552-571.
[10] Bernhoft RA.Cadmium toxicity and treatment[J].Sci World J,2013,2013:1-7.
[11] Babah,Tsuneyama K,Kumada T,etal.Histopathological analysis forosteomalacia and tubulopathy in itai-itai disease[J].J Toxicol Sci,2014,39(1): 91-96.
[12] Adiele RC,Stevens D,Kamunde C.Features of cadmium and calcium uptake and toxicity in rainbow trout (Oncorhynchusmykiss)mitochondria[J].Toxicol in Vitro,2012,26(1):164-173.
[13] Jorge-Nebert LF,Galvez-Peraltam,Landerofigueroa J,etal.Comparing gene expression during cadmium uptake and distribution:untreated versus oral Cd-treated wild-type and ZIP14 knockoutmice[J].Toxicol Sci,2014,143(1):26-35.
[14] Prozialeck WC,Edwards JR.Mechanisms of cadmium-induced proximal tubule injury:newinsights with implications for biomonitoring and therapeuticinterventions[J].J Pharmacol Exp Ther,2012,343(1):2-12.
[15]matović V,Buha A,Bulat Z,etal.Cadmium toxicity revisited:focus onoxidative stressinduction and interactions with zinc andmagnesium[J].Arch Toxicol,2011,62(1):65-75.
[16] Wang J,Zhuh,Liu X,etal.N-acetylcysteine protects against cadmium-induced oxidative stressin rathepatocytes[J].J Vet Sci,2014,15(4):485.
[17] 海春旭.自由基医学[M].西安:第四军医大学出版社,2006.
[18] Ben P,Zhang Z,Xuan C,etal.Protective effectof l-theanine on cadmium-induced apoptosisin PC12 cells by inhibiting themitochondria-mediated pathway[J].Neurochem Res,2015,40(8):1661-1670.
[19] Luevano J,Damodaran C.A review ofmolecular events of cadmium-induced carcinogenesis[J].J Environ Pathol Toxicol Oncol, 2014,33(3):183-194.
Dose-dependent inductionof renal injury by cadmium in rats
WANG Lele,LIU Jiangzheng,LIUmengmeng,KONG Deqin,ZHANG Tao,YU Weihua,LIU Rui,ZHANG Xiaodi,WANG Xin,HAI Chunxu*
(Departmentof Toxicology, School of Publichealth, Fourthmilitarymedical University, Shaanxi Key Lab of Free Radical Biology andmedicine, Xi'an 710032, Shaanxi, China)
目的:探 讨不同剂量氯化镉(CdCl2)染毒对大鼠肾损伤的量效关系及其机制。方法:雄性SD大鼠40只,随机分为0、1、3和5mg/kg CdCl2染毒组,每天灌胃1次。连续染毒4周后检测血清尿素氮(BUN)和肌酐(SCr)水平评价肾功能;HE染色观察肾脏病理损伤;透射电镜观察肾组织超微结构变化;DHE及MitoSOX荧光探针检测肾组织中活性氧(ROS)含量变化;免疫印迹检测SOD1、SOD2、GPx-1、CAT、Bax、Bcl-2以及GRP78蛋白的表达。结果:与对照组比较,镉暴露后大鼠体质量及肾体比差异无统计学意义(P> 0.05);随CdCl2剂量增加,血清BUN含量逐渐增加(r=0.463,P<0.05);肾脏病理损伤进行性加重,出现肾小球肾小管肿胀,肾小管管腔狭窄、上皮细胞坏死脱落堆积于管腔、管腔内可见管型,肾间质充血,炎细胞浸润;超微结构观察显示,肾小管上皮细胞线粒体损伤,肿胀、变形、空泡样变程度逐渐加重;肾组织中ROS含量逐渐增加;抗氧化酶SOD2、GPx-1和CAT表达逐渐增加(r依次为0.854、0.975、0.918,P均<0.05),SOD1表达逐渐减少(r=-0.987,P<0.05);肾脏组织促凋亡蛋白Bax表达在0~3m g/kg C dCl2处理组逐渐增加(r=0.226,P<0.05),抑凋亡蛋白Bcl-2表达逐渐减少(r=-0.85,P<0.05),Bax/Bcl-2呈剂量依赖性升高(r=0.83,P<0.05); 同时,内质网应激标志蛋白GRP78的表达也随CdCl2剂量增加而升高(r=0.913,P<0.05)。结论:不同剂量镉暴露致雄性SD大鼠肾脏损伤存在剂量效应关系,其作用机制可能是镉暴露导致氧化还原稳态失衡,从而引起氧化应激,进一步引起凋亡和组织损伤。
镉暴露;肾损伤;剂量效应关系;氧化应激;活性氧
OBJECTIVE:This study was designed to investigate the dose-dependent relationship and the possiblemechanisms of renal injury by cadmium in rats.METHODS:Fortymale rats were randomly divided intofour groups (0,1,3 and 5mg/kg CdCl2) groups.The rats were intra-gastrically administrated CdCl2daily to investigate the inductionof renal injury.Blood urea nitrogen (BUN) and serum creatinine (SCr) weremeasured to evaluate renal function.Kidney tissues were stained withhematoxylin-eosin (HE) and observed forhistopathological changes.Transmission electronmicroscopye was used to detect ultrastructural abnormalities.Levels of ROsin kidney was detected by DHE andmitoSOX staining.The levels of SOD1,SOD2,GPx-1,CAT,Bax,Bcl-2,GRP78 weremeasured by Western blotting.RESULTS:After exposure to CdCl2for 4 weeks,the relative kidney weights showed no significant difference among the 4 groups.As the dose of CdCl2increased,the levels of BUN elevated gradually (r=0.463,P<0.05).Pathological damages to kidneys were progressively aggravated.Swelling of glomerulus and renal tubule,and necrosis of tubular epithelial cells were observed under lightmicroscopy.And it was also observed that the lumina of renal tubules contained necrotic epithelial cellular debris and casts.Interstitialhyperemia and inflammatory infiltration were found as well.The ultrastructure ofmitochondria in the epithelium of proximal convoluted tubule showed swelling,deformation and vacuolation in a dose-dependentmanner.The level of kidney ROS elevated gradually.Levels of SOD2,GPx-1 and CAT
cadmium exposure;renal injury;dose-dependent;oxidative stress;reactive oxygen species
R994.6
A
1004-616X(2016)06-0446-07
1 0.3969/j.issn.1004-616x.2016.06.007
2016-07-19;
2016-09-28
国家自然科学基金(81473010);陕西省自然科学基金(2016JM8024)
作者信息: 王乐乐,E-mail:diana1147824730@163.com。*通信作者,海春旭,E-mail:cx-hai@fmmu.edu.cn
were upregulated (r=0.854,0.975,0.918,P all<0.05) while SOD1 were downregulated (r=-0.987,P<0.05) as the dose of CdCl2increased.Meanwhile,levels of the apoptosis-promoting Bax increased (r=0.226,P<0.05) while the antiapoptotic Bcl-2 decreased (r=-0.85,P<0.05) and the ratio of Bax/Bcl-2 increased (r=0.83,P<0.05).The levels of GRP78 also increased when the dose of CdCl2increased (r=0.913,P<0.05).CONCLUSION:The results demonstrate that there was a dose-dependent toxic effectof CdCl2on renal structure and function.Moreover,oxidative stressmight be the keymechanism involved in the toxicity of cadmium because of increased antioxidase expression and levels of ROS.
