利用CRISPR/Cas9技术制备Mindin基因敲除小鼠
2016-12-07纪慈数赵志刚
纪慈数,赵志刚,贺 颖*
(1.厦门大学实验动物中心,福建厦门361102;2.广州军区武汉总医院,湖北武汉430000)
·研究简报·
利用CRISPR/Cas9技术制备Mindin基因敲除小鼠
纪慈数1,赵志刚2,贺 颖1*
(1.厦门大学实验动物中心,福建厦门361102;2.广州军区武汉总医院,湖北武汉430000)
针对Mindin基因编码序列设计3条单链向导RNA(sgRNA),制备相应质粒,将表达sgRNA和Cas9蛋白的质粒共同电转入小鼠成纤维细胞L929中,通过药物筛选获得细胞库.经单克隆测序检测细胞库中基因组的突变效率,选择效率最佳的sgRNA.体外转录获取sg RNA和Cas9 m RNA,进行小鼠受精卵的显微注射后提取小鼠基因组DNA测序鉴定.产生的40只小鼠中17只发生不同程度的碱基插入或缺失,其中23号小鼠缺失10个碱基造成移码突变,Mindin基因表达被破坏,成功构建出Mindin基因敲除小鼠,为深入研究Mindin基因的生物学功能奠定了基础.
CRISPR/Cas9;基因敲除;Mindin
CRISPR/Cas9是近年来从古细菌免疫系统中发展起来的一种新型高效的基因组核酸内切酶编辑技术,因其操作简单、成本低、效率高、限制条件少等优点而备受关注,是目前生命医学领域的研究热点[1-3].
Mindin是一种高度保守的分泌性细胞外基质蛋白,通过介导细胞与基质之间的相互作用对细胞的黏附、分化、迁移和增殖等产生重要影响[4].之前的研究大都集中在Mindin对免疫调节的重要作用[7]上,对Mindin在肿瘤生成和发展中的作用研究较少.已有研究表明Mindin在多种人类肿瘤中异常表达[6-7].增强Mindin的表达会引起肠癌细胞迁移、侵袭和增殖能力的提升[18],提示Mindin在肠癌病理过程中发挥重要的抗癌作用.本研究拟利用CRISPR/Cas9技术显微注射C56BL/6J小鼠受精卵制备Mindin基因敲除小鼠,为深入研究Mindin基因的生物学功能奠定基础,为肠癌的研究、预防与治疗提供有力的模型支持.
1 材料和方法
1.1材料
细胞株:L929细胞株(小鼠成纤维细胞株)由厦门大学李勤喜教授实验室馈赠,用含10%(体积分数)胎牛血清(FBS,Gibco公司)的DMEM培养基(Gibco公司)于5%(体积分数)CO2恒温湿热培养箱中培养.
质粒:p UC57-sgRNA质粒载体(Addgene 51132)以及pST1374-Cas9-NLS表达质粒由上海科技大学黄行许教授实验室馈赠;p BKS质粒由厦门大学林圣彩教授实验室馈赠.
感受态细胞:大肠杆菌(Escherichia coli)DH5α感受态细胞购自Genentech公司.
1.2主要试剂与仪器
PCR仪(BioRad公司),凝胶成像系统(Genentech公司),电转仪(BioRad公司),显微注射仪(NIKON&NARISHIGE GROUP).
PCR扩增及质粒构建:BsaⅠ限制性内切酶(NEB,R0535S),SolutionⅠ(Ta KaRa,6022Q),Primerstar(Premix)(Ta KaRa,DR040A).
体外转录:MEGAshortscriptTMT7 Kit(Life Technology,AM1354),m MESSAGE m MACHINE® T7 ULTRA Kit(Life Techonology,AM1345), MEGAclearTMKit(Ambion,AM1908),A-PlusTMPoly(A)Polymerase Tailing Kit(CellScript,C-PAP5104H).体外转录依据试剂盒使用说明书进行.
单链向导RNA(sgRNA)寡聚核苷酸链及单克隆引物由厦门安特奥生物有限公司合成;质粒测序及单克隆菌液测序由厦门闽博生物有限公司完成.
1.3实验方法
1)质粒构建:针对Mindin基因中前两段蛋白编码序列,利用麻省理工大学张锋教授实验室提供的在线网站(http:∥crispr.mit.edu/)分析可能的sg RNA位点(PAM序列为NGG),从中挑选3个高评分的sgRNA位点.构建相应sgRNA表达质粒,通过测序检验所构建质粒的正确性.
2)质粒的电转及敲除效率检测:将测序正确的sgRNA质粒连同Cas9质粒共同电转入小鼠成纤维细胞L929中,加入杀稻瘟菌素(blasticidin)筛选获得细胞库;提取细胞库基因组,用相应单克隆引物PCR构建单克隆进行测序,获得细胞库敲除效率.
3)体外转录及sgRNA和Cas9 m RNA的显微注射:利用合成的带T7启动子序列的引物通过PCR从相应质粒上扩增sgRNA和Cas9序列,回收PCR产物作为体外转录的模板;利用T7体外转录试剂盒体外转录sgRNA和Cas9 mRNA,Cas9 mRNA转录完后在3′端加入poly(A)尾以增加m RNA的稳定性;最后利用试剂盒回收sgRNA和Cas9 m RNA,并稀释至终质量浓度分别为50和100 ng/μL进行受精卵显微注射(胞质注射).
4)小鼠基因组的提取及突变基因的检测:受精卵显微注射后,将其移回至假孕母鼠的输卵管内.待所生小鼠(F0代)断奶后切取尾部组织(约3 mm),酚-三氯甲烷抽提法提取基因组DNA,PCR获得相应靶基因片段并进行测序,筛选突变的小鼠.
5)F0代小鼠的传代及F1代小鼠的检测:将获得的F0代突变小鼠与野生型小鼠配对,获得F1代小鼠,提取肠上皮细胞进行蛋白质免疫印迹,检测Mindin蛋白的表达.
2 结果和分析
2.1sgRNA的设计和相应质粒的构建
利用http:∥crispr.mit.edu/网站针对Mindin基因编码序列设计挑选了3个sg RNA作用靶点(表1).加入通用连接序列合成相应寡聚核苷酸链,退火后连接入BsaⅠ切割的p UC57-sgRNA载体,完成sg RNA质粒载体的构建,并通过测序验证.
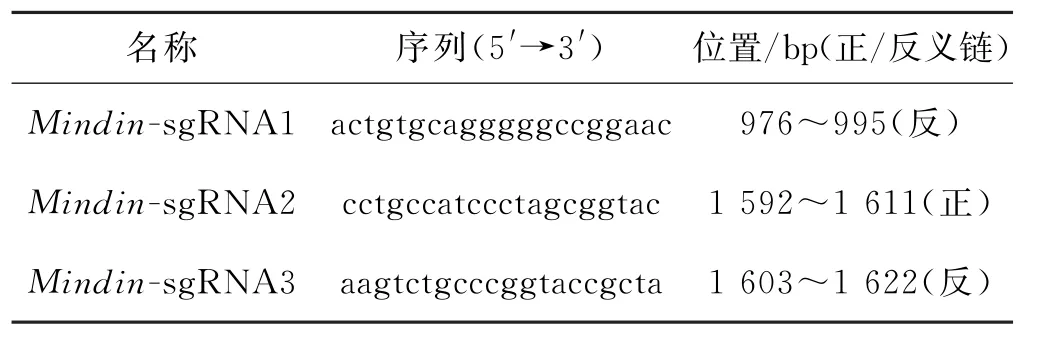
表1 针对小鼠Mindin基因编码序列的sg RNA位点信息Tab.1 Information of sg RNAs targeting coding sequence in mammalian Mindin gene
2.2sgRNA敲除效率的检测
为了确定所选sgRNA介导Mindin靶序列的编辑效率,将上述3条sg RNAs的表达质粒分别与表达Cas9蛋白的质粒共同电转入小鼠成纤维细胞L929中,以空白载体作为对照,实验组和对照组转染24 h后加入杀稻瘟菌素处理,待对照组细胞全部死亡后,将实验组剩下的细胞继续培养至24孔板中.利用酚-三氯甲烷抽提法提取细胞基因组进行PCR,所得DNA片段回收后连入pBKS质粒载体中,挑选单克隆菌落进行测序分析,统计各sgRNA的敲除效率.结果表明,所有单克隆中Mindin的靶位点序列相较于野生型Mindin序列都发生了改变,并且突变位点都位于sgRNA后NGG序列附近(如图1所示),说明这3条sgRNAs都可以介导Cas9蛋白高效切割Mindin基因靶序列.
2.3利用CRISPR/Cas9制备Mindin基因敲除小鼠
选择sg RNA3进行后续Mindin基因敲除小鼠的制备,利用带T7启动子的特异性引物PCR获得相应sg RNA和Cas9编码序列.回收的PCR产物利用T7 RNA聚合酶进行体外转录,回收RNA后按sg RNA 50 ng/μL,Cas9 mRNA 100 ng/μL的终质量浓度混合,通过胞质注射200个小鼠受精卵,最终成活160个二细胞胚胎,移植6只假孕小鼠,有5只小鼠生育,最后共获得40只F0代小鼠.
小鼠出生21 d后,切取尾部组织提取基因组DNA,用单克隆引物(表2)进行PCR.将扩增产物构建至p BKS载体中,次日挑选单克隆进行测序分析.结果如图2所示:12号小鼠的Mindin编码序列缺少了3 bp,但不影响其正常编码,为缺失突变体;23号小鼠的Mindin基因缺少了10 bp,形成移码突变,可能导致Mindin基因无法正常表达,实现了Mindin基因的敲除.

图1 sgRNA1~3细胞库的单克隆测序检测结果Fig.1 Single clone sequencing results of sgRNA1-3 cell pools

表2 sg RNA3单克隆测序引物Tab.2 sg RNA3 single clone sequencing primers
为了验证所产生的基因组突变能否稳定传代并尽量消除可能的脱靶效应,将23号小鼠与野生型小鼠进行交配,产生的F1代杂合小鼠进行基因型鉴定,测序结果如图2中23号小鼠的序列,说明Mindin基因的缺失能稳定遗传;同时提取了F1代杂合小鼠的肠上皮组织,对Mindin蛋白的表达进行蛋白质免疫印迹检测,发现F1代杂合小鼠肠组织中Mindin的表达有明显降低(图3),表明通过CRISPR/Cas9系统成功制备了Mindin基因敲除小鼠.
3 讨 论
基因组编辑是研究基因功能以及进行基因治疗的重要手段,人工核酸酶技术的出现为基因组编辑提供了强有力的工具.但早期的锌指核酸酶技术(zinc finger nuclease,ZFN)因为专利限制及锌指蛋白自身识别序列限制,应用范围不广泛[9].后期发展起来的类转录激活因子效应核酸酶(transcription activator-like effector nuclease,TALEN)技术克服了ZFN编辑技术中的识别序列限制,但其制备过程耗时而繁冗,限制了其广泛应用[10].CRISPR/Cas9作为新一代基因组编辑技术,其对靶位点的识别依赖于sg RNA 5′端约20 bp序列与靶位点DNA序列间的互补配对,针对不同靶位点序列的sgRNA仅需替换识别位点处20 bp序列即可,且其切割效率明显高于ZFN和TALEN,应用简单易行,具有显著的优势[11].
真核生物基因组DNA具有复杂高级结构,寻找高效的sg RNA位点是CRISPR/Cas9有效工作的重要保证.为了获得高效的sg RNA,在本研究中针对肠癌抑制基因Mindin的编码序列,利用已有网站设计并构建了针对Mindin编码序列的3条sgRNAs,并将其导入小鼠成纤维细胞L929中,检测sg RNA的效率.结果表明,细胞库的所有单克隆都发生了突变,并且突变位点都位于NGG序列附近,证明了这3条sgRNAs都可以介导Cas9蛋白高效切割Mindin基因靶序列.利用体外转录RNA和受精卵显微注射技术成功制备了可以稳定遗传的Mindin基因突变小鼠,其F1代杂合小鼠肠上皮细胞中Mindin蛋白的表达量显著下调,从而为深入研究Mindin在结直肠癌发生过程中的作用提供了可用的动物模型.
值得注意的是,40只F0代中总共获得了17只突变小鼠,突变率为42.5%,这与已有报道的超过70%的效率[12-13]仍有差距.之前的研究指出,体外转录RNA的质量是影响CRISPR制备基因敲除动物的关键因素[14-15].在本研究系统中,突变率低的可能原因在于显微注射过程中RNA的降解,即由于所用显微注射仪在操作过程中会发生受精卵培养基回流至注射针管导致RNA的降解,从而使效率降低;此外,Cas9 m RNA体外加poly(A)的效率也是一个可能的影响因素,poly(A)会影响CRISPR蛋白的翻译效率[16],如果体外加入poly(A)的效率偏低,也会导致效率偏低.因此,今后将从RNA质量方面进一步优化该系统.

图2 F0代突变小鼠中突变染色体的单克隆测序结果Fig.2 Single clone sequencing results of mutant chromosome in F0 mutant mouse
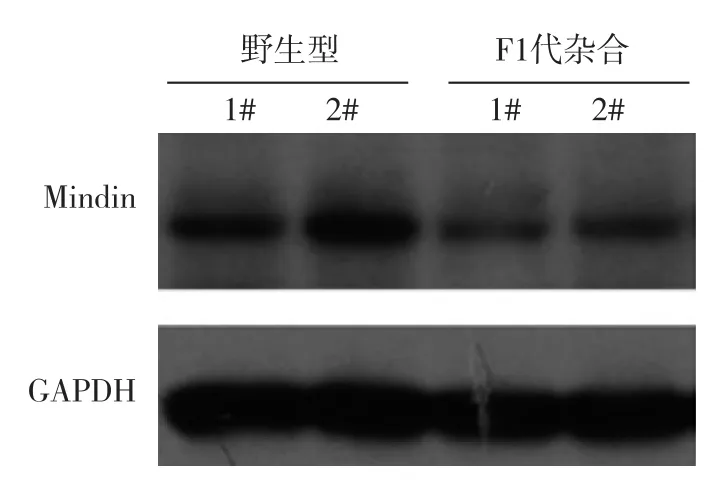
图3 F1代杂合小鼠肠上皮中Mindin表达量显著下调Fig.3 Down regulation of Mindin expression in colon epithelial cells in F1 heterozygote mice
[1] BARRANGOU R,FREMAUX C,DEVEAU H,et al. CRISPR provides acquired resistance against viruses in prokaryotes[J].Science,2007,315(5819):1709-1712.
[2] WANG H,FU Y,REYON D,et al.Efficient genome editing in zebrafish using a CRISPR-Cas system[J].Nat Biotechnol,2013,31(3):227-229.
[3] MALI P,ESVELT K M,CHURCH G M.Cas9 as a versatile tool for engineering biology[J].Nat Methods,2013, 10(10):957-963.
[4] LI Z,GARANTZIOTIS S,JIA W,et al.The extracellular matrix protein Mindin regulates trafficking of murine eosinophils into the airspace[J].J Leukoc Biol,2009,85(1): 124-131.
[5] JIA,W.The extracellular matrix protein Mindin serves as an integrin ligand and is critical for inflammatory cell recruitment[J].Blood,2005,106(12):3854-3859.
[6] LUO,J H,REN B,KERYANOV S,et al.Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas[J].Hepatology,2006,44(4): 1012-1024.
[7] MANDA R,KOHNO T,MATSUNO Y,et al. Identification of genes SPON2 and C20orf2 differentially expressed between cancerous and noncancerous lung cells by mRNA differential display[J].Genomics,1999,61(1): 5-14.
[8] BA G,LIAN Y M,REN J L.Mindin is upregulated during colitis and may activate NF-kappaB in a TLR-9 mediated manner[J].World J Gas,2010,16(9):1070-1075.
[9] GEURTS A M,COST G J,FREYVERT Y,et al. Knockout rats via embryo microinjection of zinc-finger nuclease[J].Science,2009,325(5939):433.
[10] CERMAK T,DOYLE E L,CHRISTIAN M,et al.Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting[J]. Nucleic Acids Res,2011,39(12):82-84.
[11] CONG L,RAN F A,COX D,et al.Multiplex genome engineering using CRISPR/Cas systems[J].Science,2013, 339(6121):819-823.
[12] WANG H,YANG H,SHIVALILA C S,et al.One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J].Cell, 2013,153(4):910-918.
[13] SHEN B,ZHANG J,WU H,et al.Generation of genemodified mice via Cas9/RNA-mediated gene targeting [J].Cell Res,2013,23(5):720-723.
[14] YANG H,WANG H,JAENISCH R,et al.Generating genetically modified mice using CRISPR/Cas-mediated genome engineering[J].Nat Protoc,2014,9(8): 1956-1968.
[15] QIN W,KUTNY P M,MASER R S,et al.Generating mouse models using CRISPR-Cas9-mediated genome editing[J].Curr Protoc Mouse Biol,2016,6(1):39-66.
[16] CHU V T,WEBER T,GRAF R,et al.Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes[J].BMC Biotechnol,2016,16:4.
Application of CRISPR/Cas9 Technology for Establishing Mindin Gene Knockout Mice
JI Cishu1,ZHAO Zhigang2,HE Ying1*
(1.Laboratory Animal Center,Xiamen University,Xiamen 361102,China; 2.Wuhan General Hospital,Guangzhou Military,Wuhan 430000,China)
To establish Mindin knockout mice using CRISPR/Cas9 gene targeting technology,three sg RNA targets of Mindin were designed according to Mindin genome sequence.Vectors expressing sgRNA and Cas9 were constructed separately,and co-transfected into L929 cells by with electroporation method.Transfected cells were selected with blasticitin and the mutation rate was determined using DNA sequencing with proper primers.The sgRNA which had resulted in the highest mutation rate in L929 cells was transcribed in vitro,along with Cas9.Then sgRNA and Cas9 m RNAs were microinjected into mice zygotes and 40 mice were born after the microinjection.Genotypes of the mice were determined by PCR with genomic DNA from the new born mice tail and 17 neonatal mice were found carrying mutations in Mindin gene by subsequent sequencing.Gene sequencing results showed that all mutant mice had different levels of nucleotide insertion or deletion,with the maximum number of 10 base deletion found in mouse No.23,leading to the reading frame shift and Mindin gene silence.Taking together,we have successfully established a mouse model with Mindin gene knockout,which lays a solid foundation for related future research.
CRISPR/Cas9;gene knockout;Mindin
Q 789
A
0438-0479(2016)06-0922-05
10.6043/j.issn.0438-0479.201604028
2016-04-15 录用日期:2016-06-22
福建省自然科学基金重点项目(2013Y01010490)
hey@xmu.edu.cn
纪慈数,赵志刚,贺颖.利用CRISPR/Cas9技术制备Mindin基因敲除小鼠[J].厦门大学学报(自然科学版),2016,55 (6):922-926.
JI C S,ZHAO Z G,HE Y.Application of CRISPR/Cas9 technology for establishing Mindin gene knockout mice[J]. Journal of Xiamen University(Natural Science),2016,55(6):922-926.(in Chinese)
