CE和HPLC测定水源水体中微囊藻毒素方法比较
2016-12-06徐明芳曾晓琮耿梦梦陈耕南
王 阳,徐明芳,*,曾晓琮,耿梦梦,黎 明,陈耕南
(1.暨南大学生命科学技术学院,广东 广州 510632;2.暨南大学应急管理研究中心,广东 广州 510632;3.广东省食品检验所,广东省酒类检测中心,广东 广州 510435)
CE和HPLC测定水源水体中微囊藻毒素方法比较
王 阳1,2,徐明芳1,2,*,曾晓琮3,耿梦梦1,黎 明1,陈耕南1
(1.暨南大学生命科学技术学院,广东 广州 510632;2.暨南大学应急管理研究中心,广东 广州 510632;3.广东省食品检验所,广东省酒类检测中心,广东 广州 510435)
利用毛细管电泳(capillary electrophoresis,CE)仪结合紫外-可见光二极管阵列检测器检测技术建立水体中痕量微囊藻毒素的检测新方法。通过与GB/T 20466—2006《水中微囊藻毒素的测定》高效液相色谱(high performance liquid chromatography,HPLC)方法对比分析,进行2 种检测技术的评价。CE检测条件为:毛细管柱(60 cm×75 μm i.d.),有效长度为44 cm,分离缓冲溶液为12 mmol/L硼酸盐(pH 9.0),分离电压25 kV,检测波长238 nm,压力6 895 Pa流体动力学进样;优化后的HPLC检测条件为:C18色谱柱(250 mm×4.6 mm i.d.,5 μm),甲醇-磷酸盐缓冲溶液(60∶40,V/V)为流动相,检测波长238 nm,柱温度35 ℃,流速1 mL/min。结果表明:CE对3 种微囊藻毒素MC-RR、MC-LR和MC-YR的检出限分别是0.16、0.20 μg/mL和0.24 μg/mL,HPLC对3 种微囊藻毒素的检出限分别为0.020、0.079 μg/mL和0.052 μg/mL,这2 个方法的灵敏度相差1 个数量级;加标回收率分别92.5%~106.0%和99.6%~102.5%,CE对应的保留时间和峰面积的精密度相对标准偏差为0.53%~0.64%和2.67%~3.29%;HPLC法的保留时间和峰面积精密度相对标准偏差为0.16%~0.53%和0.80%~1.53%。检测同一水样中微囊藻毒素含量,CE检测结果和HPLC结果之间差异不显著(P>0.05)。
高效毛细管电泳;高效液相色谱;微囊藻毒素;检测
微囊藻毒素(microcystins,MCs)主要是由铜绿微囊藻(Microcystis aeruginosa)产生的细胞内毒素,具有生物活性的单环七肽化合物,分子质量约为1 000 D[1],如图1所示,它在细胞内合成,细胞破裂后释放出来,导致水体中MCs的出现,能抑制控制生化过程的生物体蛋白磷酸酶,是一种强烈的肝肿瘤促进剂[2-3]。不仅直接污染饮用水源,还可以在水生生物中富集,通过食物链而进入高等生物体内,直接威胁人类的健康和生存[4-7],引起胃肠炎症,也会引发消化道炎症、肝癌和脾脏疾病等,剂量过度时可导致死亡[8]。目前,MCs、黄曲霉毒素和乙肝病毒已成为环境中致肝癌的三大危险因素[9],而且已经发现的MCs有80多种变体[10-11],包括MC-LR、MC-YR、MC-RR等,其中分布最广泛、毒性最强的是MC-LR。世界卫生组织推荐的饮水中MC-LR标准为1.0 μg/L[12],加拿大标准为0.5 μg/L[13],我国参照世界卫生组织标准,在卫生部2001年6月颁布的《生活饮用水水质卫生规范》中确定MC-LR的限值标准定为1.0 μg/L。华南地区饮用水水源地在藻华暴发期,曾检测出较高浓度的藻毒素。因此,加强水质藻毒素监控与风险评估不仅关系人们用水和身体健康的问题,也是关系到公共安全与社会稳定能否持续发展的关键问题之一,建立快速、先进、可靠和高效的藻毒素检测技术,完善水中MCs的测定方法至关重要。
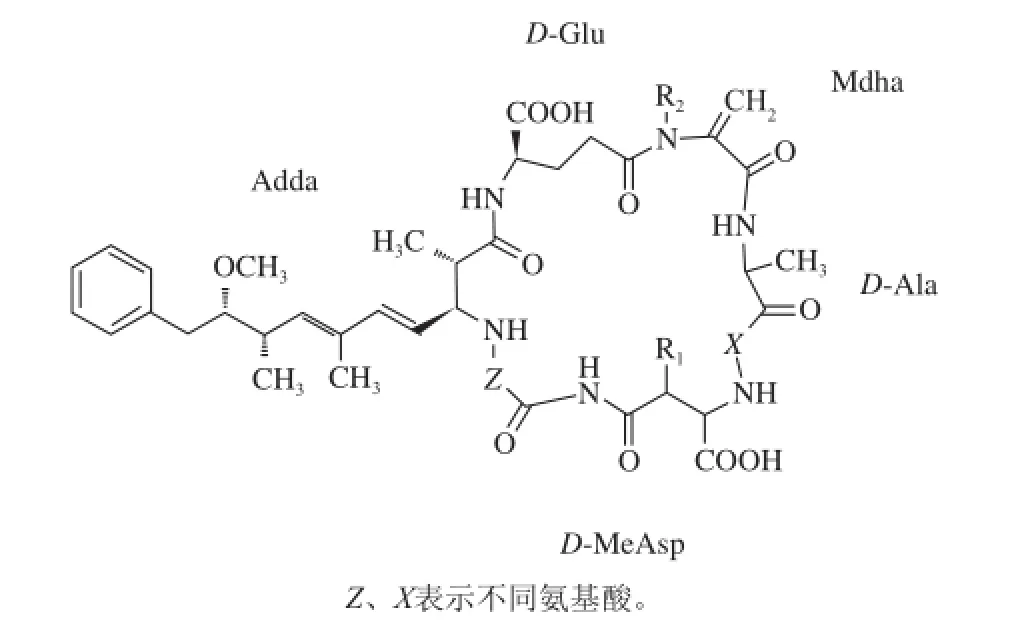
图1 MCs分子结构图Fig.1 General structures of microcystins
目前,MCs测定方法主要包括高效液相色谱(high performance liquid chromatography,HPLC)[14]、气相色谱、HPLC-质谱(HPLC-mass spectrometry,HPLC-MS)联用[15]、2-甲基-3-甲氧基-4-苯基丁酸法[16]和酶联免疫吸附(enzyme linked immuno sorbent assay,ELISA)法[17]。ELISA方法不仅可检测MCs的各种异构体及检测样品中的蓝藻毒素总量,而且样品也不需要复杂的前处理就可直接进行检测,经济、快速,特别适合于应对突发公共卫生事件的处理,及缺乏大型仪器设备的基层进行调查研究,但是该方法易出现假阳性[18]。HPLC-MS对样品前处理要求不高,可以同时分离和鉴定多种藻类毒素,而且具有更高的选择性和灵敏度。但HPLC-MS联用仪器昂贵且运行成本较高,一般的实验室没有能力购买和负担,所以不适合普及和广泛使用。毛细管电泳(capillary electrophoresis,CE)法是最新的色谱分离技术,它根据分子的大小以及所带电荷来分离样品,不仅满足了仪器分析所要求的快速、高效、样品用量少等特点,而且具有操作简单、仪器自动化程度高、对环境污染小等优点[19-20]。而HPLC法是目前对毒素进行分析最常用的技术之一,该方法分析速度较快,能准确地定性、定量检测MCs。本实验利用CE仪结合紫外-可见光二极管阵列检测器建立水体中藻毒素检测新方法,通过与GB/T 20466—2006《水中微囊藻毒素的测定》中HPLC方法学的对比,进行2 种方法学的评价,为水中MCs高效测定新方法提供理论依据。
1 材料与方法
1.1 材料与试剂
MCs标准品:MC-LR(纯度≥98%,CAS 101043-37-2)、MC-RR(纯度≥98%,CAS 111755-37-4)Taiwan Algal Science Inc.;MC-YR(纯度≥98%,CAS 101064-48-6) 瑞士Enzo公司;甲醇、三氟乙酸(均为色谱纯) 天津市科密欧化学试剂有限公司;四硼酸钠、硼酸、磷酸、磷酸二氢钾(均为分析纯) 广州化学试剂厂。
1.2 仪器与设备
Coulter P/ACETM MDQ高效CE仪(配自动进样器和
二极管阵列紫外检测器) 美国Beckman公司;338454型毛细管柱(60 cm×75 μm i.d.,有效长度44 cm)美国Sciex公司;1100 HPLC仪(数据分析平台附带荧光检测器)、C18色谱柱(250 mm×4.6 mm i.d.,5 μm)美国Agilent公司;500 mg/6 mL C18固相萃取小柱 广州特克斯科学仪器有限公司。
1.3 方法
1.3.1 标准溶液的配制
MCs标准储备液:分别称取MCs标准品MC-LR、MC-RR、MC-YR各100 μg,用0.1 mL甲醇溶液溶解,再用纯水定容到10 mL,放在棕色试剂瓶中-20 ℃保存30 d。此标准储备液的质量浓度约为10 μg/mL。
系列标准溶液:用体积分数20%甲醇溶液将MCs标准储备液分别稀释至所需质量浓度(临用时配制)。
1.3.2 缓冲溶液的配制
硼砂电泳缓冲液:称取适量的四硼酸钠,溶于超纯水中,配制成12 mmol/L四硼酸钠溶液,用1 mol/L硼酸溶液调其pH值为9.0。
磷酸盐缓冲溶液:用体积分数20%磷酸溶液将磷酸二氢钾溶液(0.05 mol/L)调节pH 3.0。
1.3.3 样品的前处理
水样的采集和保存:用采水器采集1 500~2 000 mL水样,在水下0.5 m处进行采样(水样采集后,应在4 h内完成前处理步骤)。用500 目的不锈钢筛过滤,除去水样中大部分浮游生物和悬浮物。取过滤的水样1 200 mL于玻璃杯式滤器中,依次经过GF/C玻璃纤维滤膜和0.45 μm乙酸纤维酯滤膜减压过滤,准确量取1 000 mL滤液置于棕色试剂瓶中。
样品的富集、洗脱:依次用10 mL体积分数100%甲醇溶液和10~15 mL纯水活化C18固相萃取小柱→滤液(控制流速为8~10 mL/min)→依次用淋洗溶液(10 mL水、10 mL体积分数20%甲醇溶液)淋洗C18固相萃取小柱→10 mL洗脱溶液(用甲醇将0.1 mL三氟乙酸溶液定容到100 mL)洗脱MCs→洗脱液在40 ℃条件下用旋转蒸发器浓缩吹干,用0.5 mL体积分数100%甲醇溶液溶解以供测定用。
1.3.4 分离条件
CE分离条件:毛细管柱温25 ℃;检测波长238 nm;压力6 895 Pa;进样时间5 s;运行电压25 kV。
HPLC分离条件:C18色谱柱(250 mm×4.6 mm i.d.,5 μm);柱温度35 ℃;流动相:甲醇-磷酸盐缓冲溶液(60∶40,V/V)混合;流速1 mL/min;紫外-可见光检测器波长238 nm;进样量50 μL。
1.3.5 样品的测定及计算
利用上述2 种方法分别测定珠江西航道水中MCs含量,比较2 种分析方法对于样品的适用性。
水样中MCs含量(X1)按下式计算:
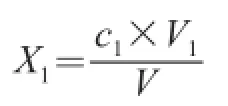
式中:X1分别代表MC-RR、MC-YR、MC-LR质量浓度/(μg/L);c1为从标准曲线上查出的MCs(MC-RR、MC-YR、MC-LR)含量/(μg/mL);V1为水样定容体积/mL;V为采集水样的体积/L。
1.4 数据处理
每个水样平均测定3 次,用统计软件SPSS 13.0计算结果,用Origin 7.5进行绘图。
2 结果与分析
2.1 CE分离条件的选择
2.1.1 缓冲液浓度对分离结果的影响
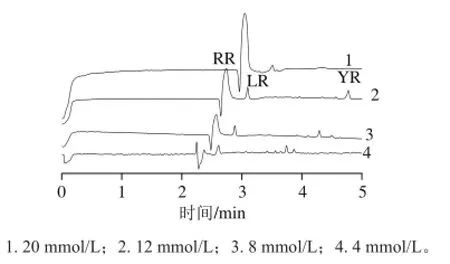
图2 不同浓度硼酸盐缓冲溶液的MCs电泳图Fig.2 Electropherogram of MCs standards with borax buffer solutions of different concentrations
硼酸盐浓度的改变明显影响着溶质的迁移时间,进而影响电渗流的大小及分析物质的有效电荷,从而决定其在毛细管中的迁移速率,所以选择合适的硼酸盐浓度可以改变分析物质的分离选择性[21]和迁移行为。如图2所示,在其他条件不变的情况下,检测不同浓度的硼酸盐缓冲液(4、8、12、20 mmol/L)对毛细管电泳分离MCs标准物质效果的影响,以便找到最佳的缓冲液浓度。当使用高浓度缓冲液(20 mmol/L)时,出现了电流增大、峰不全以及迁移时间延长的现象。使用低浓度缓冲液(4 mmol/L和8 mmol/L)时,虽然迁移时间缩短,但检测噪音增大,导致MCs检出限的降低。使用12 mmol/L的硼酸盐溶液可以提高电泳的选择性和分辨率,同时峰形更尖锐,电泳分离效率更高。所以12 mmol/L的硼酸盐缓冲液是CE分离MCs的最佳缓冲液浓度。
2.1.2 缓冲溶液pH值对分离结果的影响
pH值的高低会引起溶质所带电荷的改变[22],影响溶质的迁移行为。pH值的改变会引起电渗流的变化、分离情况的变化,所以缓冲溶液的pH值选择对MCs的分离尤为重要。本实验采用了4 个pH值(8.7、9.0、9.52、10.05)。如图3所示,pH值为10.05和9.52时,目标物不
能完全分离;当pH值为8.7时,峰形更变宽,电泳分离效率降低,而且基线也不稳定。使用pH值为9.0,目标物不仅得到了完全分离,而且目标物的峰形尖锐,电流也比较稳定。所以pH值为9.0的12 mmol/L硼酸盐缓冲液是CE分离MCs的最佳缓冲液pH值。
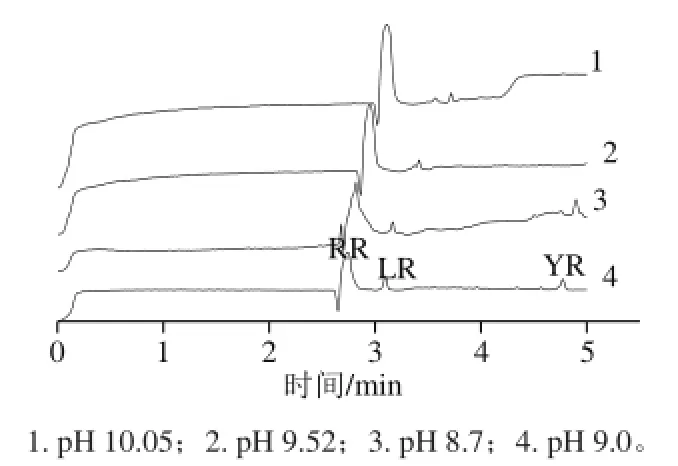
图3 不同pH值条件下的电泳图Fig.3 Electropherograms of MCs standards under different pH values
2.1.3 运行电压对分离结果的影响
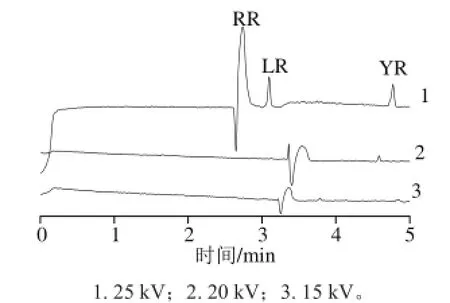
图4 不同电压条件下的电泳图Fig.4 Electropherogram of MCs standards under different voltages
如图4所示,分离电压会影响分离速度和多组分间的分离度。当分离电压为30 kV时,电泳分离过程中产生的焦耳热高。运行电解质容易产生气泡,电泳过程的电流不稳定,基线噪声大[23]。当分离电压为20 kV和15 kV时,分离度不高,影响对MCs的识别。综合实验结果,运行电压定为25 kV最合适。
2.1.4 MCs混合标准溶液测定
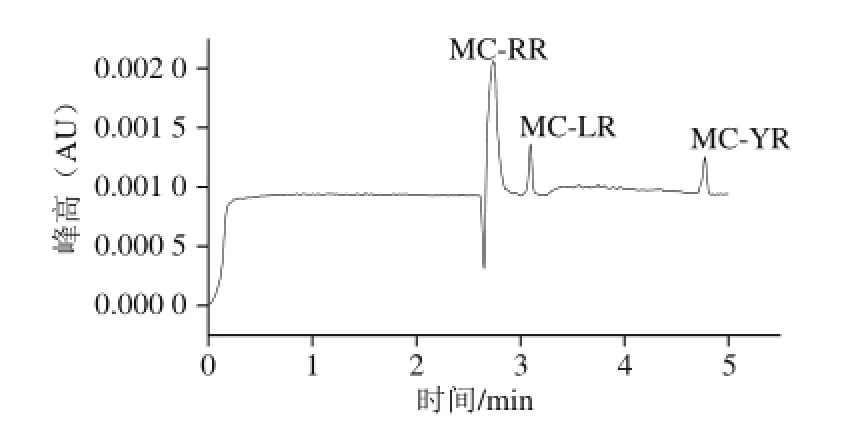
图5 MCs混合标准溶液CE图Fig.5 CE electropherogram of MCs standards
如图5所示,通过实验得到的CE的分离条件为pH 9.0 的12 mmol/L硼酸盐缓冲液,运行电压为25 kV,色谱峰峰形较好、分离度较高。
2.2 HPLC分离条件的优化
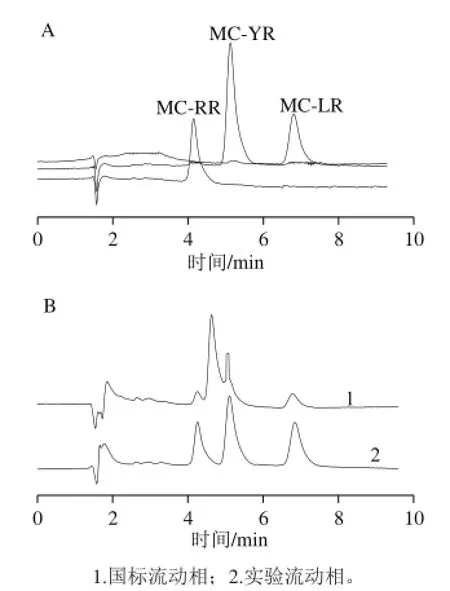
图6 MCs单标(A)和混合标准溶液(B)HPLC图Fig.6 HPLC profiles of MCs standards
采用GB/T 20466—2006《水中微囊藻毒素的测定》HPLC法,如图6A及图6B中谱线1所示,甲醇和磷酸盐缓冲溶液的体积比为57∶43时,发现目标峰与杂质峰有部分重叠,分离不彻底。在HPLC检测的过程中,流动相配比[24]对峰形和出峰时间均有一定的影响。因此,为确保MCs的最优分离度和出峰时间,选择甲醇和磷酸盐缓冲溶液的体积比为60∶40,如图6A及图6B中谱线2所示,在谱线1基础上,增加甲醇在流动相所占的比例。因此在国标的基础上,对分离条件进行相应的优化,各物质的保留时间变快,而且图谱基线平稳,峰形好,且与杂质能实现较好分离,峰的顺序(出峰时间)分别为MC-RR (4.2 min)、MC-YR(5.1 min)、MC-LR(6.8 min),所有样品在7 min内检测完毕。
2.3 回归方程、线性范围和检出限比较
分别用HPLC法和CE法测定MCs的标准溶液,以质量浓度为横坐标,对应的峰面积为纵坐标,绘制标准曲线,拟合回归方程及相关系数(R2)。以3 倍信噪比所对应的质量浓度计算检出限,如表1所示。
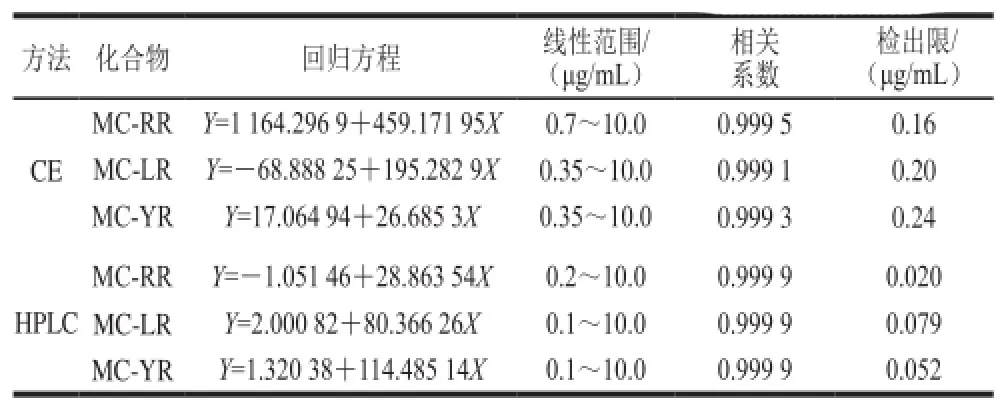
表1 MCs的标准曲线、线性范围和检出限Table1 Standard curves, linearity ranges, and limits of detection (LOD) of MCs
由表1可知,2 种测定方法的线性范围、相关系数和检出限均能满足饮用水中MCs的测定要求。HPLC的检出限比CE的检出限低,可能是由于CE采用几至几十微米内径的毛细管为分离通道,所以在使用紫外、二极管阵列等光吸收为基础的检测器时,其灵敏度比较低[25]。
2.4 回收率和精密度比较
同时配制5 份相同质量浓度的标准MCs混合溶液在各实验条件下分别进样,以保留时间和峰面积为目标分析,计算各检测技术中MCs的保留时间和峰面积的相对标准差,分析仪器的精密度;并采用混合标准储备液为4 μg/mL的添加量加标,按照测量值和真实值计算回收率,考察方法的回收率,见表2。
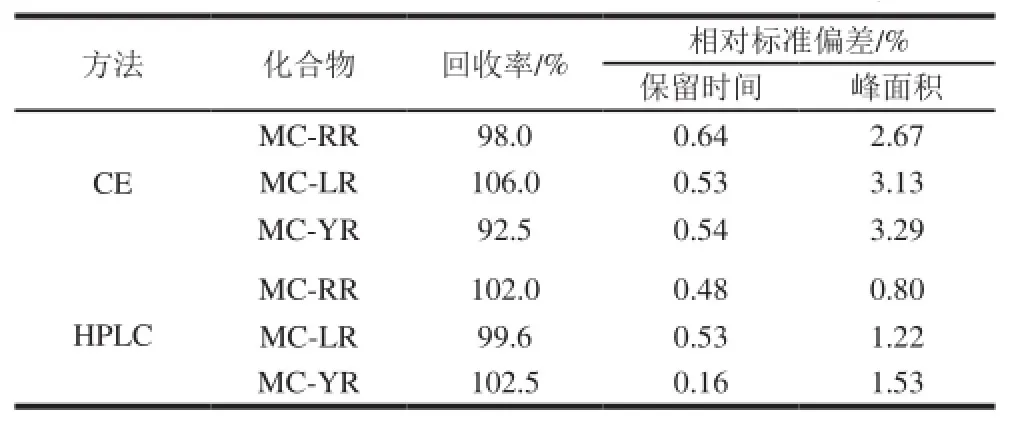
表2 水样中MCs的加标回收率(n=5)Table2 Recovery rates of spiked water samples (n= 5)
由表2可知,对2 种检测技术精密度的比较发现三组分的保留时间的相对标准偏差均小于1%,峰面积的相对标准偏差均小于5%,所以二者精密度均符合测定要求。CE法和HPLC法相比,其相同之处在于都是高效分离技术,仪器操作均可自动化,且二者均有多种不同分离模式。二者之间的差异在于CE用迁移时间取代HPLC中的保留时间,然而实验发现,CE检测法的重复性(精密度)略低于HPLC法,这是因为CE检测法的重复性受毛细管柱与电场电压影响很大,因为在CE中电泳分析的整个过程,当毛细管两端施加高电压时,高电场强导致电流增加,引起毛细管中电解质产生焦耳热(自热)。自热将使流体在径向产生抛物线型温度分布,即管轴中心温度要比近壁处温度高。因溶液黏度随温度升高呈指数下降,温度梯度使介质黏度在径向产生梯度,从而影响溶质迁移速度,使管轴中心的溶质分子要比近管壁的分子迁移得更快,造成柱效下降,尤其是高电场时产生流向阴极的电渗流,而目标分析物的迁移速度则是电泳和电渗流速度的矢量和,毛细管内壁存在对缓冲液的吸附性,影响电渗流,继而影响分离的重现性[23]。因此毛细管柱与检测器不断发展是CE用于毒素检测更广泛应用的重要基础。
2.5 2 种方法测定水样中MCs含量比较
采用CE和HPLC方法,对采集的珠江西航道水样中MCs的含量进行测定,平均每个样品测定3 次,如表3所示。
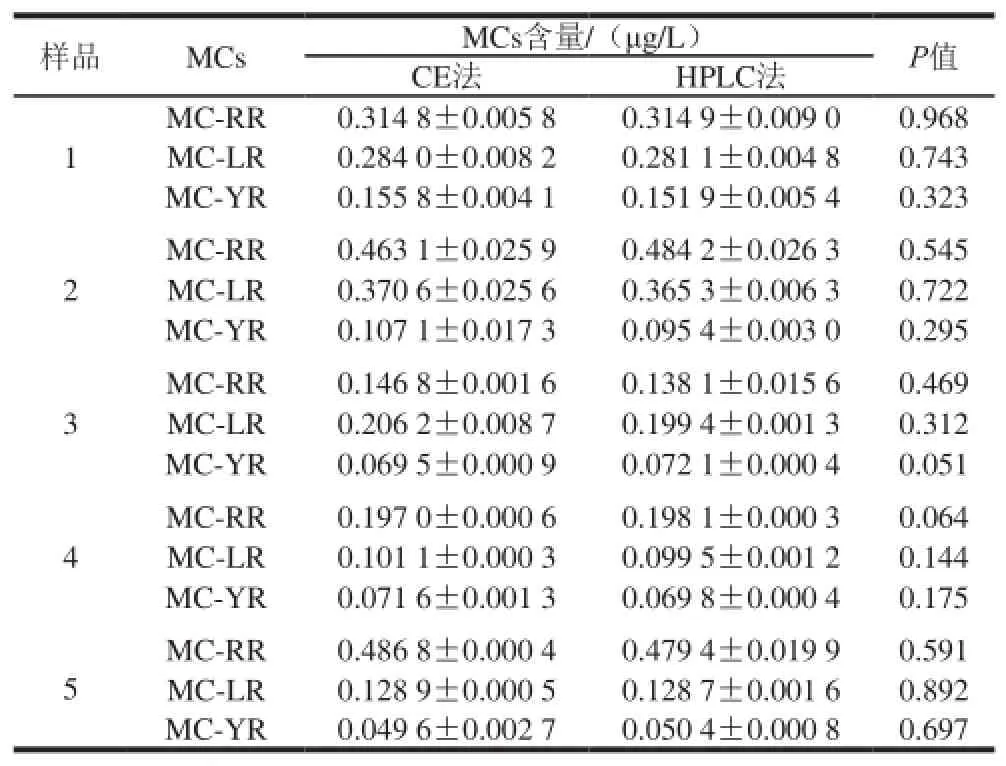
表3 水样中MCs的含量Table3 The contents of MCs in water samples from the Pearl River
由于天然水体中MCs含量比较低,所以要进行MCs的检测时,首先必须按照1.3.3节方法进行处理,对MCs进行富集。然后按照检测条件进行测定。CE和HPLC 2 种方法的检测结果都低于国家标准(1 μg/L)。但是由于这些样品都是在丰水期采集的,水体流动性比较大,MCs含量会比较低,而且这些MCs会在水生生物体内富集,然后通过食物链进入人体进而影响健康,所以珠江西航道水中MCs还是存在相当大的隐患。对CE法和HPLC法测定结果之间进行检验,2 种检测结果差异不显著(P>0.05);相同的检测样品,由于CE技术采用高电场,具有分离效率高、受基质干扰小等特点,所以检测结果中只有相对应的3 种MCs,方便观察和计算(图7A);从图7B可以看出,含有很多的杂质,而且各个色谱峰之间的分离不彻底,出峰时间延后,存在连带现象,可能是水体中的杂质干扰或者分离程序选择的不好。
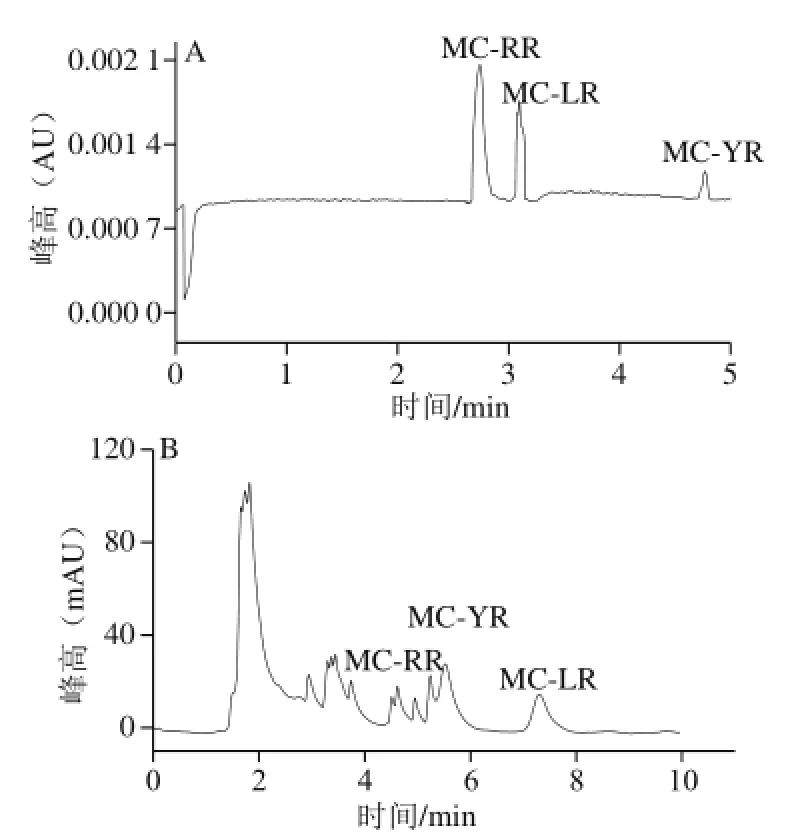
图7 水样中MCs CE(A)和HPLC(B)图Fig.7 CE (A) and HPLC (B) profiles of MCs in water sample from the Pearl River
3 结 论
在GB/T 20466—2006《水中微囊藻毒素的测定》方法基础上,优化水中MCs含量的HPLC检测条件,结果表明MCs回收率为99.6%~102.5%,相对标准偏差为0.80%~1.53%,对MC-LR的检出限为0.079 μg/mL,但是HPLC法分析易受到样品中其他物质的干扰。
建立水中MCs含量的CE检测方法,结果表明,MCs回收率为92.5%~106.0%,相对标准偏差为2.67%~3.29%,对MC-LR检出限为0.20 μg/mL。同时CE技术具有分离效率高、分析速度快的优点。
本实验通过测定珠江西航道水中MCs的含量,对CE技术和HPLC法2 组测定结果进行了对比分析,结果显示两者之间差异性不显著(P>0.05),定量结果相一致。所以具有分析时间短、需要的样品量少、有机试剂消耗少、分析成本低、污染小、受基质干扰小等优势的CE方法可以用于MCs含量检测,并为水中MCs高效、快速测定新方法的提供理论依据。
[1] ANTONIOU M, CRUZ A, DIONYSIOU D. Cyanotoxins: new generation of water contaminants[J]. Journal of Environmental Engineering, 2005, 131(9): 1239-1243. DOI:10.1061/(ASCE)0733-9372(2005)131:9(1239).
[2] BORMANS M, LENGRONNE M, BRIENT L, et al. Cylindrospermopsin accumulation and release by the benthic cyanobacterium Oscillatoria sp. PCC 6506 under different light conditions and growth phases[J]. Bulletin of Environmental Contamination and Toxicology, 2014, 92(2): 243-247. DOI:10.1007/ s00128-013-1144-y.
[3] 许川, 舒为群. 微囊藻毒素污染状况、检测及其毒效应[J]. 国外医学(卫生学分册), 2005, 32(1): 56-60. DOI:1001-1226(2005)01-0056-05.
[4] DAI R, WANG P, JIA P, et al. A review on factors affecting microcystins production by algae in aquatic environments[J]. World Journal of Microbiology and Biotrchnolgy, 2016, 32(3): 1-7. DOI:10.1007/s11274-015-2003-2.
[5] LIU Y, CHEN W, LI D, et al. Cyanobacteria-/cyanotoxincontaminations and eutrophication status before Wuxi Drinking Water Crisis in Lake Taihu, China[J]. Journal of Environmental Sciences, 2011, 23(4): 575-581. DOI:10.1016%2fS1001-07-42(10)60450-0.
[6] ZHANG D, PING X, CHEN J. Effects of temperature on the stability of microcystins in muscle of fish and its consequences for food safety[J]. Bulletin of Environmental Contamination and Toxicology, 2010, 84(2): 202-207. DOI:10.1007/s00128-009-9910-6.
[7] 谢平. 微囊藻毒素对人类健康影响相关研究回顾[J]. 湖泊科学, 2009, 21(5): 603-613. DOI:10.3321/j.issn:1003-5427.2009.05.001.
[8] 俞顺章, 赵宁, 资晓林, 等. 饮水中微囊藻毒素与我国原发性肝癌关系的研究[J]. 复旦学报(医学科学版), 2001, 23(2): 96-99. DOI:10.3760/j.issn:0253-3766.2001.02002.
[9] 熊筱璐, 韩晓冬, 曾莉. 微囊藻毒素毒性机制研究进展[J]. 中国公共卫生, 2015, 31(2): 238-241. DOI:10.11847/zgggws2015-31-02-32.
[10] SHARMA V K, TRIANTIS T M, ANTONIOU M G, et al. Destruction of microcystins by conventional and advanced oxidation processes: a review[J]. Separation and Purification Technology, 2012, 91(3): 3-17. DOI:10.1016/j.seppur.2012.02.018.
[11] 王小宁, 杨玉玺, 宗万松. 微囊藻毒素生物毒性作用机制与调控策略的研究进展[J]. 环境污染与防治, 2015, 37(6): 90-93. DOI:10.15985/j.cnki.1001-3865.2015.06.017.
[12] GORCHEV H G, OZOLINS G. WHO guidelines for drinking-water quality[M]//World Health Organization, 2004: 104-108.
[13] Federal-Provincial Committee on Environmental and Occupational Health. Guidelines for canadian drinking water quality[S].
[14] 中国科学院水生生物研究所. 水中微囊藻毒素的测定: GB/T 20466—2006[S]. 北京: 中国标准出版社, 2006.
[15] 李秋霞, 刘辉, 蔡超海, 等. 超高效液相色谱-串联质谱法同时检测生活饮用水中7 种微囊藻毒素[J]. 华南预防医学, 2015, 41(6): 593-595. DOI:10.13217/j.scjpm.2015.0593.
[16] 聂晶晶, 李元, 李琴, 等. 微囊藻毒素检测方法的研究进展[J]. 中国环境监测, 2007, 23(2): 43-48. DOI:10.3969/j.issn.1002-6002.2007.02.012.
[17] 谭瑶, 舒为群, 邱志群. 生物样本中微囊藻毒素检测方法的研究进展[J]. 环境卫生学杂志, 2014, 4(1): 93-98. DOI:10.13421/j.cnki. hjwsxzz.2014.01.016.
[18] 杨振宇. 水和水产品中微囊藻毒素的检测方法研究[D]. 上海: 复旦大学, 2010: 9-12.
[19] 李津津, 黄燕, 杨德草. 毛细管电泳技术在中药研究方面的应用情况分析[J]. 当代医药论丛, 2014, 12(4): 147-149. DOI:10.3969/ j.issn.2095-7629.2014.04.124.
[20] 王百木, 刘昌云. 毛细管电泳技术在视频检测中的应用[J]. 中国调味品, 2011, 36(7): 24-29. DOI:10.3969/j.issn.1000-9973.2011.07.007.
[21] 邓延倬, 何金兰. 高效毛细管电泳[M]. 北京: 科学出版社, 1996: 124-127.
[22] 蒋晓颖. 用于环境分析的毛细管电泳方法及应用研究[D]. 上海:华东师范大学, 2014: 26-38.
[23] 欧婉露, 李玉娟, 石冬冬, 等. 非水毛细管电泳法测定藤黄中藤黄酸的含量[J]. 色谱, 2015, 33(2): 152-157. DOI:10.3724/SP.J.1123.2014.11006.
[24] 孙睿华. 微囊藻毒素的高效液相色谱测定法若干问题的探讨[J]. 环境与健康杂志, 2015, 32(5): 448-449. DOI:10.16241/ j.cnki.1001-5914.2015.05.021.
[25] ALNBIN M, GROSSMAN P D, MORING S E. Sensitivity enhancement for capillary electrophoresis[J]. Analytical Chemistry. 1993, 65(10): 489A-497A. DOI:10.1021/ac00058a720.
Comparison of HPLC and CE for Estimation of Microcystins in Drinking Water Sources
WANG Yang1,2, XU Mingfang1,2,*, ZENG Xiaocong3, GENG Mengmeng1, LI Ming1, CHEN Gengnan1
(1. College of Life Science and Technology, Jinan University, Guangzhou 510632, China; 2. Research Center of Emergency Management, Jinan University, Guangzhou 510632, China; 3. Guangdong Provincial Institute of Food Inspection, Guangdong Provincial Wine Testing Center, Guangzhou 510435, China)
A new method for the detection of trace amounts of microcystins (MCs) in drinking water sources by capillary electrophoresis (CE) coupled with ultraviolet/visible light diode array detector (DAD) system was established and compared with the high performance liquid chromatography (HPLC) described in the Chinese standard method (GB/T 20466—2006). CE detection conditions were determined as follows: an uncoated fused-silica capillary tube (60 cm × 75 μm i.d.) with effective length of 44 cm as stationary phase, 12 mmol/L sodium borate solution (pH 9.0) as running buffer, separation voltage of 25 kV, sample injection under 6 895 Pa for 5 s, and detection wavelength of 238 nm. The HPLC method was performed with a C18chromatographic column (250 mm × 4.6 mm i.d., 5 μm) using methanol: phosphate buffer solution (60:40, V/V) as mobile phase at a flow rate of 1 mL/min. The column temperature was set at 35 ℃, and the analyte was detected at 238 nm. The limits of detection (LOD) of the CE method for MC-RR, MC-LR and MC-YR were 0.16, 0.20 and 0.24 μg/mL, and those of the HPLC method were 0.020, 0.079, and 0.052 μg/mL, respectively. The sensitivity of the two methods differed by 1 order of magnitude. The recoveries of the CE and HPLC methods for three MCs were in range of 92.5%–106.0% and 99.6%–102.5%, respectively. The corresponding relative standard deviations (RSDs) of retention time and peak area were 0.53%–0.64% and 2.67%–3.29% for CE and 0.16%–0.53% and 0.80%–1.53% for HPLC, respectively. For the same water sample, the MCs content determined by CE was not significantly different from that determined by HPLC (P > 0.05).
capillary electrophoresis (CE); high performance liquid chromatography (HPLC); microcystins (MCs); detection
10.7506/spkx1002-6630-201622032
X824
A
1002-6630(2016)22-0210-06
王阳, 徐明芳, 曾晓琮, 等. CE和HPLC测定水源水体中微囊藻毒素方法比较[J]. 食品科学, 2016, 37(22): 210-215. DOI:10.7506/spkx1002-6630-201622032. http://www.spkx.net.cn
WANG Yang, XU Mingfang, ZENG Xiaocong, et al. Comparison of HPLC and CE for estimation of microcystins in drinking water sources[J]. Food Science, 2016, 37(22): 210-215. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201622032. http://www.spkx.net.cn
2016-05-26
暨南大学应急管理研究中心重大项目(JD2015008);广东省科技计划项目(2009B011300003)作者简介:王阳(1991—),女,硕士研究生,研究方向为饮用水中微囊藻毒素检测以及安全标准。
E-mail:wangyang5221@126.com
*通信作者:徐明芳(1962—),女,教授,博士,研究方向为食品特性鉴定与分离技术、生物反应器工程。
E-mail:xumingfang@jnu.edu.cn
