播散性浅表性光化性汗孔角化症SLC17A9基因突变分析
2016-12-02杨付希安田洪青刘红张福仁
夏 杨付希安田洪青刘 红张福仁
·短篇论著·
播散性浅表性光化性汗孔角化症SLC17A9基因突变分析
夏 杨1,2付希安2田洪青2刘 红2张福仁2
目的: 检测11例山东汉族播散性浅表性光化性汗孔角化症SLC17A9基因突变位点。方法: 提取患者外周血DNA,采用PCR扩增患者SLC17A9基因的全部外显子及其侧翼序列,对PCR扩增产物直接测序检测。结果: 11例播散性浅表性光化性汗孔角化症(DSAP)患者的SLC17A9基因编码区的所有外显子均未发现突变。结论: 本研究中11例DSAP患者的发病与SLC17A9基因的编码区序列无关。
播散性浅表性光化性汗孔角化症; SLC17A9基因; 基因突变
汗孔角化症(porokeratosis,PK)是一种常染色体显性遗传的慢性进行性角化性皮肤病,具有遗传异质性[1]。本病的发病诱因有遗传、免疫抑制、紫外线、感染及外伤等。该病是由 Mibelli于 1893年首次报道[2],根据形态学、皮损分布、临床特征的不同,目前公认分为5种临床类型[3]:Mibelli型汗孔角化症(PM),播散性浅表性汗孔角化症(DSP),播散性浅表性光化性汗孔角化症(DSAP),掌跖合并播散性汗孔角化症(PPPD),线状汗孔角化症(LP)。其中,播散性浅表性光化性汗孔角化症(Disseminated superficial actinic porokeratosis,DSAP)是一组以环状排列的异常角化性皮损为特征表现,主要发生在曝光部位的常染色体显性遗传性皮肤病,它是汗孔角化症中最为常见的一种类型,30~40岁多见,由Chenosky等于1969年在德克萨斯人群首先描述[4],DSAP作为一种遗传性疾病,其表型改变是以基因的改变为基础,因此寻找其致病基因不仅可以对疾病做出明确诊断,而且可以在分子水平更好的认识其发病机制。通过传统的以家系为基础的全基因组连锁分析研究已经发现了6个DSAP的致病区域[5-10]:12q23.2-24.1,12q24.1-q24.2,15q25.1-26.1,Ip31.3-p31.1,16q24.1-24.3和20q13.33。2014年,张学军团队通过对1个DSAP家系进行全外显子测序发现新的致病基因SLC17A9,并在6家系和19个散发DSAP患者中进行验证,其中,在DSAP-2家系中发现SLC17A9基因突变位点1个[10]。为进一步验证SLC17A9为 DSAP的致病基因,我们对收集到的4例DSAP家系的先证者及7例散发共11例患者进行SLC17A9基因突变位点检测。
1 资料与方法
1.1 研究对象 本研究选择的用于验证的DSAP样本都是经之前MVK基因检测未发现变异的样本,从中选择4例有家族史的DSAP患者以及7例散发DSAP病例进行基因检测(图1、2)。
1.2 材料 外周血DNA的提取:核对临床资料、临床图片及组织病理检查结果,从-80℃冰柜取出冷冻保存的血液,采用AXYGEN试剂盒提取基因组DNA。取适量DNA进行纯度检测并标化。
1.3 方法
1.3.1 PCR扩增 通过美国国家生物信息中心(NCBI)基因库及UCSC数据库检索到SLC17A9基因的基因组序列,采用Premier Primer5.0软件设计合适的引物,由北京华大基因科技有限公司合成(表1)。按照说明书将其稀释为20μmol/L,并采用温度梯度法探索每一对引物的最佳退火温度。PCR反应在9700型PCR扩增仪(美国ABI公司)上完成,反应条件为: 94℃预变性3min,94℃变性30 s,退火45 s,72℃延伸45 s,共35个循环。最后72℃延伸8 min,4℃保存。
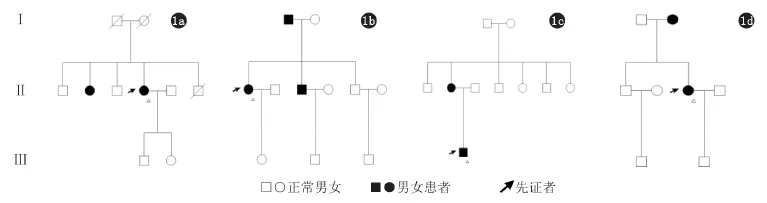
图1 家系图(1a:家系1;1b:家系2;1c:家系3;1d:家系4。箭头示进行基因测序检测的患者)
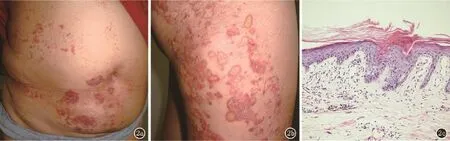
图2 2a:散发1患者腹部典型的DSAP皮损:曝光部位分布的边缘堤状隆起,中央轻度萎缩的褐色皮疹2b:散发1患者右侧臀部和大腿的典型皮损2c:典型的角化不全柱(HE,×200)

表1 本研究中设计的引物
1.3.2 DNA测序 PCR产物采用ABI3130xl遗传分析仪(美国ABI公司)直接测序。
2 结果
直接测序结果SLC17A9基因整个编码区具有编码功能的13条外显子及其侧翼序列被扩增,并且个别外显子采用了双向测序,以确保测序结果的准确性。11例患者的DNA测序结果中均未发现突变位点。
3 讨论
DSAP是汗孔角化症最常见的一种类型,属常染色体显性遗传。暴露于紫外线和辐射治疗为本病的诱发因素。长时间日晒后皮损可加重,因此皮损主要分布于面、颈、躯干、四肢伸侧等曝光部位,表现为离心性角化性丘疹,界清,中央轻度凹陷萎缩,边缘有突起的嵴。本病的诊断依靠皮损特点、临床表现及组织病理学检查。
到目前为止,通过全基因组扫描技术、单倍型和重组连锁分析等方法共定位了DSAP的6个致病区域:12q23.2-24.1,12q24.1-q24.2,15q25.1-26.1,1p31.3-p31.1,16q24.1-24.3和20q13.33[5-10]。并在上述 6个致病区域中,发现 4个致病基因 SSH1 (12q24.1-q24.2)[11],SART3(12q24.1-q24.2)[6],MVK(12q24)[12]和 SLC17A9(20q13.33)[10],在其他致病区域未发现致病基因。
有证据表明SLC17A9负责溶酶体ATP积累[13]。据说ATP可激活蛋白酶如溶酶体中的组织蛋白酶D[14]和增加蛋白质水解。组织蛋白酶D缺乏可引起溶酶体脂褐质累积[15]表明,SLC17A9调节溶酶体蛋白水解作用通过保持ATP水平足以激活蛋白酶,如组织蛋白酶D。因此,没有功能的SLC17A9可以减弱组织蛋白酶D活动,促进脂褐质的积累和细胞死亡[13]。
SLC17A9是一种活化的ATP转运蛋白,它编码蛋白感受不同的刺激因素启动ATP的转运,如无机盐离子,蛋白激酶 C等。其中无机盐离子中包括Ca2+,K+,Cl-以及 Br-等[16],负电荷无机盐离子依赖的SLC17A9在结构上与Cl-依赖的VGLUT相似。外来因素的刺激可以通过不同的通路来激活离子浓度的改变,从而启动由SLC17A9介导的ATP的转运,比如在T细胞受体受抗原刺激可以激活蛋白激酶C,导致细胞外Ca2+流入,从而启动ATP转运。与ATP转运伴随发生的细胞内Ca2+浓度变化对于维持表皮的分化至关重要,表皮屏障功能缺失内质网中的Ca2+的丢失可以导致增高水平的细胞增殖和降低水平的分化标记的表达,同样Ca2+浓度增加可以增加分化水平,抑制增殖[17]。对于角质形成细胞,Ca2+通过质膜向细胞内的运输对细胞分化进程有至关重要的作用[18]。Ca2+浓度升高可以增加细胞分化水平,同时抑制其增殖。研究证实遗传性的Ca2+浓度失衡可以导致以多发性角质性丘疹为主要皮损特征的常染色体显性遗传性毛囊角化病(Darier病)[19]。与Darier病类似,DSAP同样表现角化过度,棘层肥厚以及角化不良的病理学特点,文献中推断SLC17A9基因变异导致的VNUT与ATP结合[13],转运以及释放异常,同时依赖ATP转运的Ca2+浓度发生改变,对角质形成细胞的增殖和分化产生影响,继而导致DSAP发病。
迄今已发现2个DSAP相关的SLC17A9基因突变位点(表2),但本研究中11例患者所有SLC17A9基因编码区域及侧翼序列均未发现突变。推测可能的原因:一是突变位点可能位于内含子区域,而我们并没有检测所有的内含子区域;二是该病可能存在其他致病基因。下一步我们会对非编码区进行测序,检测内含子区域是否存在突变。

表2 文献中报道的关于DSAP的致病基因SLC17A9突变位点
[1]Mibelli V.Contribute allo studio della ipercheratosi dei canali sudoriferi(porocheratosis)[J].G Ital Mai Ven,1893,28: 313-355.
[2]Wade TR,Ackerman AB.Cornoid lamellation.A histologic reaction pattern[J].Am JDermatopathol,1980,2(1):5-15.
[3]Cengizlier R,Hücümenoĝlu S,Ozen A,et al.Treatment of telangiectasia macularis eruptiva perstans with montelukast [J].Allergol Immunopathol,2009,37(6):334-336.
[4]Anderson DE,Chernosky ME.Disseminated superficial actinic porokeratosis.Genetic aspects[J].Arch Dermatol,1969,99(4):408-412.
[5]Xia JH,Yang YF,Deng H,et al.Identification of a locus for disseminated superficial actinic porokeratosis at chromosome 12q23.2-24.1[J].J Invest Dermatol,2000,144(6):1071-1074.
[6]Zhang ZH,Niu ZM,Yuan WT,et al.A mutation in SART3 gene in a Chinese pedigree with disseminated superficial actinic porokeratosis[J].Br J Dermatol,2005,152(4):658-663.
[7]Xia K,Deng H,Xia JH,et al.A novel locus(DSAP2)for disseminated superficial actinic porokeratosis maps to chromosome 15q25.1-26.1[J].Br JDermatol,2002,147(4): 650-654.
[8]Liu P,Zhang S,Yao Q,et al.Identification of a genetic locus for autosomal dominant dissem inated superficial actinic porokeratosis on chromosome 1p31.3-p31.1[J].Hum Genet,2008,123(5):507-513.
[9]Luan J,Niu Z,Zhang J,et al.A novel locus for disseminated superficial actinic porokeratosis maps to chromosome 16q24.1-24.3[J].Hum Genet,2011,129(3):329-334.
[10]Cui H,Li H,Wang W,et al.Exome sequencing identifies SLC17A9 pathogenic gene in two Chinese pedigrees with dissem inated superficial actinic porokeratosis[J].J Med Genet,2014,51(10):699-704.
[11]Zhang Z,Niu Z,Yuan W,et al.Finemapping and identification of a candidate gene SSH1 in disseminated superficial actinic porokeratosis[J].Hum Mutat,2004,24(5):438.
[12]Zhang SQ,Jiang T,Li M,et al.Exome sequencing identifies MVK mutations in disseminated superficial actinic porokeratosis[J].Nature Genetics,2012,44(10):1156-1160.
[13]Cao Q,Zhao K,Zhong XZ,et al.SLC17A9 Protein Functions as a Lysosomal ATP Transporter and Regulates CellViability[J].JBiol Chem,2014,89(33):23189-23199.
[14]Pillai S,Zull JE.Effects of ATP,vanadate,and molybdate on cathepsin D-catalyzed proteolysis[J].J Biol Chem,1985,260(14):8384-8389.
[15]Koike M,Nakanishi H,Saftig P,et al.Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNSneurons[J].JNeurosci,2000,20(18):6898-6906.
[16]Isidor B,Lindenbaum P,Pichon O,et al.Truncatingmutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis[J].Nat Genet,2011,43(4):306-308.
[17]Boyden ED,Campos-Xavier AB,Kalamajski S,et al.Recurrent dominant mutations affecting two adjacent residues in themotor domain of the monomeric kinesin KIF22 result in skeletal dysplasia and joint laxity[J].Am JHum Genet,2011,89(6):767-772.
[18]Hong JH,Yang YM,Kim HS,et al.Markers of squamous cell carcinoma in sarco/endoplasmic reticulum Ca2+ATPase 2 heterozygotemice keratinocytes[J].Prog BiophysMol Biol,2010,103(1):81-87.
[19]Ahn W,Lee MG,Kim HW,et al.Multiple effects of SERCA2b mutations associated with Darier's disease[J].JBiol Chem,2003,278(23):20795-20801. (收稿:2016-02-01 修回:2016-07-27)
·病例报告·
Analysis of SLC17A9 genemutations in dissem inated superficial actinic porokeratosis
XIA Yang1,2,FU Xi'an2,TIAN Hongqing2,LIU Hong2,ZHANG Furen2.
1.School ofMedicine and Life Sciences,University of Jinan-Shandong Academy of Medical Sciences,Jinan 250062,China;2.Shandong Provincial Institute ofDermatology and Venereology,Jinan 250022,China
Objective:To identifymutations of SLC17A9 gene in eleven patientswith disseminated superficial actinic porokeratosis.M ethods:After extracting DNA from peripheral blood,all the exons of SLC17A9 gene and their flanking intronic sequences were amplified by PCR,and then direct sequencing was performed to screen themutations in the gene.Results:No mutation was found in any of the exons in SLC17A9 gene from the eleven patients.Conclusion:The pathogenesis of the eleven patients with disseminated superficial actinic porokeratosis is not associated with the sequence of coding region in SLC17A9 gene.
disseminated superficial actinic porokeratosis;SLC17A9 gene;mutation
1济南大学山东省医学科学院医学与生命科学学院,济南,250000 2山东省皮肤病性病防治研究所,济南,250022
张福仁,E-mail:zhangfuren@hotmail.com
