先天性全身性脂肪营养不良BSCL2基因突变1例并文献复习
2016-11-26陈瑞敏
袁 欣 陈瑞敏 王 剑 张 莹
·论著·
先天性全身性脂肪营养不良BSCL2基因突变1例并文献复习
袁 欣1陈瑞敏1王 剑2张 莹1
目的 报告1例先天性全身性脂肪营养不良(CGL)患儿的临床特点及随访情况,提高对该病的认识。方法 分析1例CGL患儿病史、实验室检查和5年随访资料,对先证者及其父母行基因检测,系统复习国内外文献报告的CGL病例,归纳临床表型。结果 男,5岁11个月,因“腹胀、消瘦5年余”就诊。患儿足月顺产,出生无窒息抢救史,5月龄会抬头,1岁会扶走。患儿1月龄渐出现腹胀、消瘦,2月龄渐出现皮下脂肪消失,肌肉渐发达,3~4月龄渐出现全身皮肤色素沉着,以颈部和腋下显著,5~6月龄渐出现全身皮肤毛发增多、增粗。查体:神清,空双颊,全身皮下脂肪消失,肌肉发达,四肢静脉血管显露。全身皮肤偏黑,多毛,颈部、腋下黑棘皮(+++),皮肤弹性略差,心、肺查体未见异常,肝右肋下可扪及8 cm,质地中等。神经系统查体未见异常。智力测试72。双侧睾丸3 mL,阴茎5 cm×1.8 cm,阴毛Tanner 2期。父母体健,非近亲婚配。家族中无类似疾病患者。临床诊断先天性CGL,嘱低脂、高碳水化合物饮食。口服葡萄糖耐量试验提示糖耐量异常,予饮食控制。患儿BSCL2基因(NM_032667.6)存在:①错义突变c.713G>A, p.Gly238Asp(杂合);②碱基重复c.782dupG, p.Ile262Hisfs*12(杂合);其父亲携带错义突变c.713G>A, p.Gly238Asp(杂合),母亲携带碱基重复c.782dupG, p.Ile262Hisfs*12(杂合)。经系统检索有10篇文献中的17例CGL患儿进入文献汇总分析,其临床主要特征:全身皮下脂肪消失、肌肉发达、皮下静脉显露、肢端肥大、多毛、黑棘皮症、高胰岛素血症、高甘油三酯血症、肝脾大、脂肪肝、肝功能异常和心肌病等。结论 CGL罕见,易合并代谢性疾病。全身脂肪消失的患儿应首先考虑本病,基因确诊后应密切随访其代谢状况。本例患儿BSCL2基因携带的突变位点之一c.713G>A, p.Gly238Asp为首次报道。
先天性全身性脂肪营养不良; seipin; 基因检测; 高脂血症; 黑棘皮
1 病例资料
男,5岁11个月。因“腹胀、消瘦5年余”就诊于福建省福州儿童医院(我院)。患儿系G2P2顺产,母孕3月先兆流产,出生无窒息抢救史,出生体重3 300 g,出生身长不详。5月龄会抬头,1岁会扶走。患儿1月龄渐出现腹胀、消瘦,当地医院查肝功能异常,血串联质谱及尿有机酸检查未见异常。2月龄渐出现皮下脂肪消失,肌肉渐发达,3~4月龄渐出现全身皮肤色素沉着,以颈部和腋下显著,5~6月龄渐出现全身皮肤毛发增多、增粗。
查体:神清,消瘦外观,特殊面容 ,空双颊,全身皮下脂肪消失,肌肉发达,四肢静脉血管显露(图1)。全身皮肤偏黑,多毛,颈部、腋下黑棘皮(+++),皮肤弹性略差,心、肺查体未见异常,腹部膨隆,肝右肋下可扪及8 cm,质地中等。腹部移动性浊音阴性。神经系统查体未见异常。智力测试72(中国修订韦氏幼儿智力量表)。父母体健,非近亲婚配。家族中无类似疾病患者。表1显示患儿体格测量指标、超声和实验室检查结果。

图1 患儿正面半身照
临床诊断:先天性全身性脂肪营养不良(CGL),肝肿大,肝功能异常,双肾肿大,高甘油三酯血症,高胆固醇血症。嘱低脂、高碳水化合物饮食,定期复诊,家长诉患儿2~3岁渐出现脾气暴躁,易激惹,随访至今,患儿5岁11月,双侧睾丸3 mL,阴茎5 cm×1.8 cm,阴毛Tanner 2期。心脏彩超检查未见异常。口服葡萄糖耐量试验(OGTT)提示糖耐量异常,嘱饮食控制。

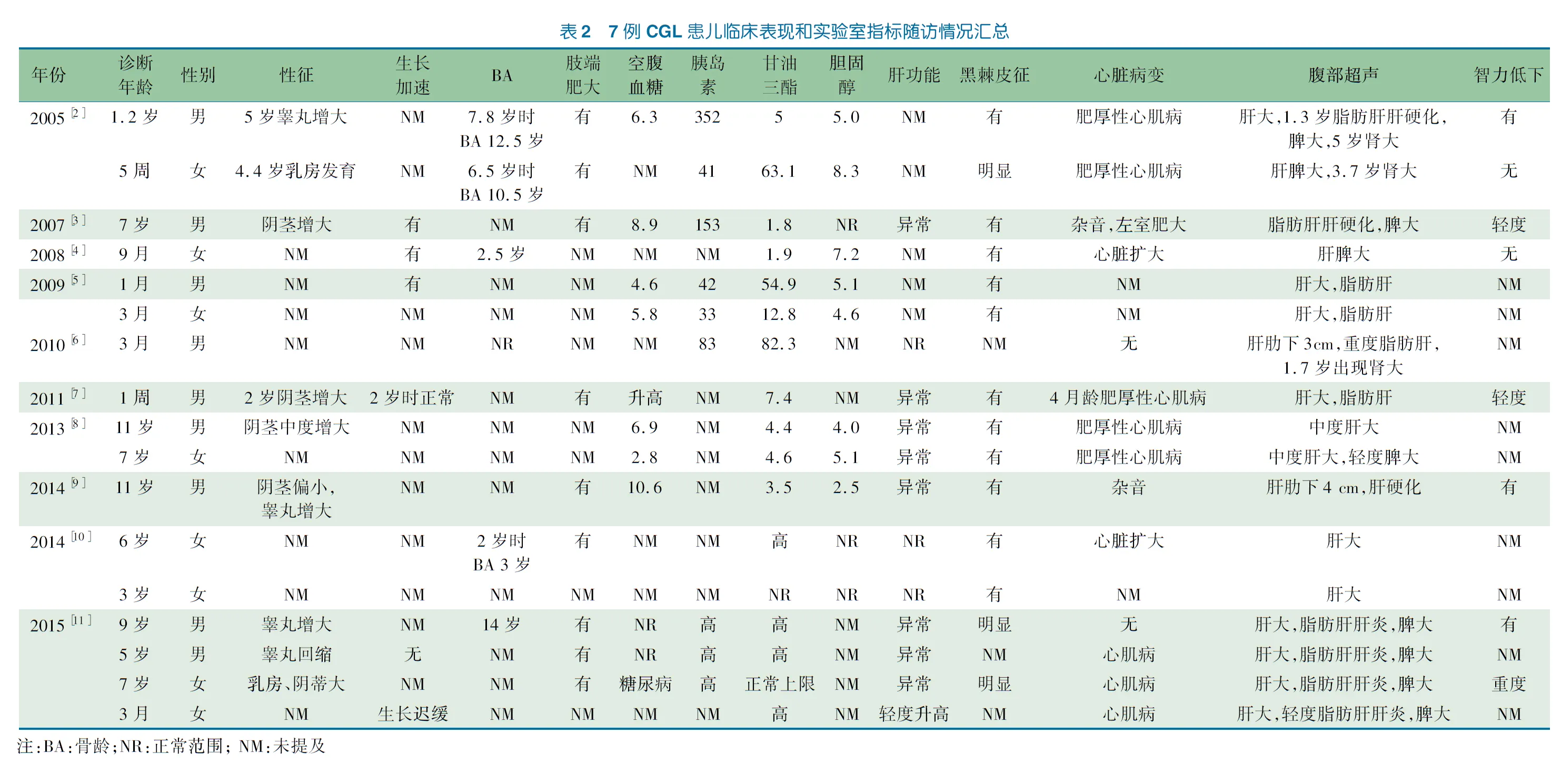
表1 患儿体格测量指标、超声和实验室检查结果
在患儿父母知情同意的情况下,EDTA抗凝管采集患儿及父母外周静脉血各5 mL,采用Agilent Sureselect目标序列富集试剂盒(美国Agilent公司)制备测序文库,捕获探针为Agilent Inherited Disease Panel(包括AGPAT2、BSCL2、CAV1和PTRF等CGL相关基因在内的2 742个遗传性疾病基因)。使用Bioanalyzer 2200仪器(美国Agilent公司)对文库的质量及浓度进行检测,试剂为High Sensitivity D1K ScreenTape和High Sensitivity D1K Reagent(美国Agilent公司)。再按照各文库样本的浓度及测序深度要求混合捕获文库,应用HiSeq2500测序仪(美国Illumina公司)进行上机测序。测序平台产生的原始数据Fastq文件经NextGENe生物信息学软件(美国SoftGenetics公司)分析后,评估测序质量,并生成VCF格式文件(包含所有变异信息)。然后将文件上传到Ingenuity在线软件系统网站(http://www.ingenuity.com/products/login)(美国 Ingenuity公司),运用“Variant Analysis”在线系统进行变异解释。最后采用Sanger测序法对候选变异进行验证。
检测到变异7 212个,经筛选流程筛选(图2),结合患儿临床资料和生物信息学软件预测结果,发现本例患儿BSCL2基因(NM_032667.6)存在:①错义突变c.713G>A, p.Gly238Asp(杂合);②碱基重复c.782dupG, p.Ile262Hisfs*12(杂合);其父亲携带错义突变c.713G>A, p.Gly238Asp(杂合),母亲携带碱基重复c.782dupG, p.Ile262Hisfs*12(杂合)(图3)。

图2 测序相关数据筛选流程图

图3 患儿及其父母BSCL2基因突变位点
注 患儿BSCL2基因存在错义突变c.713G>A, p.Gly238Asp(杂合),碱基重复c.782dupG, p.Ile262Hisfs*12(杂合);患儿父亲BSCL2基因存在错义突变c.713G>A, p.Gly238Asp(杂合);患儿母亲BSCL2基因存在碱基重复c.782dupG, p.Ile262Hisfs*12(杂合)
2 文献复习
以“先天性全身脂肪营养不良” AND “BSCL2” 为关键词在中国知网、万方数据库及中国生物医学文献数据库检索相关中文文献,未检索到基因确诊的BSCL2基因突变致CGL病例报道。
以“congenital generalized lipodystrophy” AND “BSCL2”为关键词在PubMed数据库进行检索,检索到72篇文献,排除非临床病例报道及临床资料不全的文献,并除外1例成年患者,共10篇文献[1~10]17例CGL患儿进入汇总分析,其临床主要特征见表2。
3 讨论
脂肪营养不良分为遗传性和获得性。遗传性脂肪营养不良分为家族性部分脂肪营养不良(FPLD)和CGL。CGL是一类罕见的常染色体隐性遗传病,发病率约为1/1 000万[11]。目前已知4个基因突变可导致CGL,分别是1-脂酰甘油-3-磷酸-0脂酰转酶(AGPAT2)、BSCL2、小凹蛋白1(CAV1)和聚合酶I和转录本释放因子(PTRF),分别可导致CGL1-4型。
文献复习的17例CGL患儿及本文报道患儿均表现为生后不久即发现全身皮下脂肪消失,肌肉发达,四肢静脉血管显露、特殊面容、甲状腺功能正常的高代谢状态,肝脏肿大及高甘油三酯血症等特征性临床表现,脂肪肝(11/18)及肝功能异常(9/18)为常见临床表现,出现时间较其他类型CGL更早,且程度更为严重,少数患儿早期即可发展为肝硬化[12]。此外,包括本文报道患儿在内的部分患儿尚表现出生殖器增大(9/18)、智力低下(6/18)、骨龄提前(5/18)、双肾肿大(4/18)、生长加速(4/18)等,生长加速及生殖器增大可能与高胰岛素血症直接作用于胰岛素受体或间接作用于胰岛素样生长因子1(IGF-1)受体,而产生促生长效应有关[11],肝脾肿大、肾肿大与脂质脏器沉积有关。本病需与以下引起脂肪消失的疾病相鉴别[13]:①获得性全身性脂肪营养不良,包括指膜炎相关性、自身免疫性及特发性,亦可见于HIV感染,患儿出生时脂肪分布正常,多于幼儿期发生皮下脂肪消失,而内脏脂肪不受影响,与本例患儿不符。②新生儿早老症,系FBN1 和CAV1基因突变所致,亦可出现全身性脂肪消失,但臀部及手足常不受累,且不伴有代谢性并发症。本文报道患儿与之不符,且基因诊断可排除。③非典型早老综合征,系LMNA基因突变所致。患儿常有早老的表现,如身材矮小、钩形鼻、白发、局部脱发、声音高亢和手足皮肤萎缩等。本文报道患儿无早老的相关表现,且基因诊断可排除。基因检测显示BSCL2基因突变,CGL诊断明确。
Magré等[14]于2001年首次报道BSCL2基因突变可致CGL2。CGL2是CGL中最严重的类型,缺失几乎全部具有代谢活性的脂肪组织和机械脂肪组织,智力障碍及早期死亡的发生率最高[15]。BSCL2基因位于11q13染色体,编码seipin,seipin是一个2次跨膜的内质网固有蛋白,由389个氨基酸组成,在脂肪组织、神经系统及睾丸高表达[16]。Seipin在CGL2发病机制中参与脂肪细胞分化、脂滴形成,维持脂滴形态,并限制脂滴在非脂肪细胞合成及沉积[18]。目前在CGL2患者已报道30余种BSCL2 基因突变,其中约75%为无义突变,25%为错义突变[13]。本文报道患儿BSCL2基因携带双重杂合突变c.713G>A, p.Gly238Asp; c.782dupG, p.Ile262Hisfs*12,其父母各携带1 种突变,表型正常。CGL2为常染色体隐性遗传病,一般文献报道纯合突变患儿具有该病相应的临床表现,而杂合子表型正常,BSCL2基因双重杂合突变致CGL2的病例罕见报道[10],推测双重杂合突变可能使seipin 蛋白活性下降而发病,其确切机制尚不明确。本文报道患儿携带的双重杂合突变之一c.713G>A, p.Gly238Asp为新发现突变,经Alamut功能软件预测有可能影响蛋白结构域的功能,c.713G>A(p.Gly238Asp)根据美国医学遗传学和基因组学协会(ACMGG)的指南可被判定为可能致病性,证据如下:①中等证据1(PM2):新发变异,在正常人数据库(ExAC, 1000Genome)未见该位点的频率;②中等证据2(PM3):第一个位点(c.782dupG)为已知致病位点,且该患儿BSCL2基因的两个位点各遗传自父母一方;③支持性的证据 (PP3):经SIFT、PolyPhen2以及MutationTaster在线软件预测该位点所在氨基酸位置高度保守,该突变对蛋白功能有害;④支持性的证据(PP4):患儿临床症状与该基因突变所致疾病高度吻合。另一杂合突变c.782dupG, p.Ile262Hisfs*12,已有文献报道该突变可使氨基酸翻译提前终止,形成截断型蛋白而导致CGL2[18]。
由于seipin功能异常,患儿无处理和储存能量的能力,低脂、高碳水化合物饮食是CGL2最重要的治疗手段[19]。严重高甘油三酯血症的患儿可采用苯氧酸类药物,非高密度脂蛋白胆固醇水平升高的患儿可加用低剂量他汀类药物。合并糖尿病者二甲双胍及磺脲类药物为一线用药。部分患儿需要极大剂量胰岛素才能控制血糖[20,21]。新近,美国国立卫生研究院推荐美曲普汀(重组人瘦素类似物)0.06~0.24 mg·kg-1·d-1皮下注射可有效改善胰岛素敏感性,降低甘油三酯,减少心、肝脂肪沉积,治疗后糖化血红蛋白可降低2.2%,甘油三酯水平可降低60.7%,最常见的不良反应是低血糖、头痛、恶心、降低体重和腹痛[13]。
CGL预后不良,多死于肝硬化引起食道静脉曲张破裂,肝肾功能衰竭及心脏骤停等[13]。本课题组曾报道2例临床诊断CGL的患儿[22],其中1例已死于肝功能衰竭。文献报道部分患儿合并心肌病(12/18)、高胰岛素血症(9/18)、脾肿大(8/18)及糖尿病(5/18),约45%的患儿可在青春期发生胰岛素抵抗型糖尿病[18]。本文报道患儿随访5年,经饮食控制甘油三酯有所改善,未出现心脏病变、脾肿大及肝硬化等并发症,但出现糖耐量异常,其代谢异常较文献报道病例相对更轻,提示CGL具有临床异质性,是否与其突变类型为双重杂合突变有关,尚需进一步蛋白功能研究及密切随访观察。
[1]Raygada M, Rennert O. Congenital generalized lipodystrophy: profile of the disease and gender differences in two siblings. Clin Genet 2005: 67: 98-101
[2]Jin J, Cao L, Zhao Z, et al. Novel BSCL2 gene mutation E189X in Chinese congenital generalized lipodystrophy child with early onset diabetes mellitus. Eur J Endocrinol, 2007,157(6):783-787
[3]Shirwalkar HU, Patel ZM, Magre J, et al. Congenitalgeneralized lipodystrophy in an Indian patient with a novel mutation in BSCL2 gene. J Inherit Metab Dis, 2008,31(S2):317-322
[4]Nishiyama A, Yagi M, Awano H, et al. Two Japanese infants with congenital generalized lipodystrophy due to BSCL2 mutations. Pediatr Int, 2009,51(6):775-779
[5]Huang HH, Chen TH, Hsiao HP, et al. A Taiwanese boy with congenital generalized lipodystrophy caused by homozygous Ile262fs mutation in the BSCL2 gene. Kaohsiung J Med Sci, 2010,26(11):615-620
[6]Jeninga EH, de Vroede M, Hamers N, et al. A Patient with Congenital Generalized Lipodystrophy Due To a Novel Mutation in BSCL2: Indications for Secondary Mitochondrial Dysfunction. JIMD Rep, 2012,4:47-54
[7]Rahman OU, Khawar N, Khan MA, et al. Deletion mutation in BSCL2 gene underlies congenital generalized lipodystrophy in a Pakistani family. Diagn Pathol, 2013,8:78
[8]Senanayake MP, Karunaratne I. Two unusual features in a child with Berardinelli-Seip congenital generalised lipodystrophy. Ceylon Med J, 2014,59(3):103-105
[9]Rao TS, Chennamsetty K. Berardinelli-Seip congenital lipodystrophy in two siblings. Indian Dermatol Online J, 2014,5(S1):20-22
[10]Haghighi A, Kavehmanesh Z, Haghighi A, et al. Congenital generalized lipodystrophy: identification of novel variants and expansion of clinical spectrum. Clin Genet. 2015 Jun 15
[11]Garg A. Acquired and inherited lipodystrophies. N Engl J Med, 2004 18,350(12):1220-1234
[12]Akinci B, Onay H, Demir T, et al. Natural History of Congenital Generalized Lipodystrophy: A Nationwide Study From Turkey. J Clin Endocrinol Metab, 2016,101(7):2759-2767
[13]Patni N, Garg A. Congenital generalized lipodystrophies--new insights into metabolic dysfunction. Nat Rev Endocrinol, 2015,11(9):522-534
[14]Magré J, Delépine M, Khallouf E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet, 2001,28(4):365-370
[15]Haque WA, Shimomura I, Matsuzawa Y, et al. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab, 2002,87(5):2395
[16]Lundin C, Nordstr?m R, Wagner K, et al. Membrane topology of the human seipin protein. FEBS Lett, 2006,580(9):2281-2284
[17]Wee K, Yang W, Sugii S, et al. Towards a mechanistic understanding of lipodystrophy and seipin functions. Biosci Rep, 2014,34(5). pii: e00141
[18]Agarwal AK, Simha V, Oral EA, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab, 2003,88(10):4840-4847
[19]Chan JL, Oral EA. Clinical classification and treatment of congenital and acquired lipodystrophy. Endocr Pract, 2010,16(2):310-323
[20]Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med, 2002,346(8):570-578
[21]Diker-Cohen T, Cochran E, Gorden P, et al. Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab, 2015,100(5):1802-1810
[22]陈瑞敏,陈皓,郭依华,等.全身性脂肪营养不良二例报道.中国优生与遗传杂志,2002,10(4):124
(本文编辑:张萍)
Mutation analysis of Berardinelli-Seip congenital lipodystrophy 2 gene in a patient with congenital generalized lipodystrophy and literature review
YUAN Xin1, CHEN Rui-min1, WANG Jian2, ZHANG Ying1
(1 Department of Endocrinology, Fuzhou Children's Hospital of Fujian Province, Fujian Medical University Teaching Hospital, Fuzhou 350005, China; 2 Shanghai Children's Medical Center affiliated to Shanghai Jiaotong University School of Medicine, Shanghai 200127, China)
CHEN Rui-min,E-mail: chenrm321@sina.com
Objective To analyze the clinical characteristics and the follow-up situation of a patient with congenital generalized lipodystrophy (CGL) carryingBSCL2 mutation, and to draw attention to the phenotype and genotype of patients with CGL.MethodsPhysical examination, laboratory tests, ultrasonic examination and the 5-year follow-up situation of the patient with CGL was collected, 2 742 genes of inherited diseases including CGL related genes of the patient and his parents were sequenced, and relevant literatures aboutBSCL2 mutations were reviewed.ResultsThe 5-year and 11-months boy was presented with blotting, poor weight gain for 11 months. He was full term delivered vaginally with no perinatal complications. He could raise his head at 5-month old and could walk with support at 1-year old. He appeared blot and "thin" since 1-month of age, generalized lack of body fat with extreme muscularity since 2-month of age, skin pigmentation especially in neck and armpit at 3-4 months of age, and body hair grew gradually at 5-6 months of age. Physical examination: he had abnormal face with empty cheeks, generalized lipoatrophy with absence of adipose tissue, hypertrophy of all limb muscles with prominent superficial veins, and generalized skin pigmentation especially in neck and armpit. Abdomen examination showed distension, hepatomegaly, without splenomegaly or abdominal shifting dullness. There was no special of cardiopulmonary or neurological examination. Wechsler Intelligence Scale for Children Test revised for Chinese of the patient was 72. Perineal examination showed a macropenis with 5 cm in length, testes were 3 mL in bilateral and pubic hair was Tanner 2. No similar condition was found in other members of his family. He was diagnosed as CGL and treated with low-fat, high-carbohydrate diet, oral glucose tolerance test showed impaired glucose tolerance and he was treated by diet.BSCL2 gene of the patient showed a compound heterozygous mutation: missense mutation c.713G> A, p.Gly238Asp from paternal; nucleotide repeat c.782dupG, pIle262Hisfs* 12 from maternal. A literature review of the clinical phenotypes of CGL due toBSCL2 gene mutation was performed, which showed generalized lipoatrophy with absence of adipose tissue, hypertrophy of all limb muscles with prominent superficial veins, acromegaly, hairy, skin pigmentation, hyperinsulinemiathe, hypertriglyceridaemia hepatomegaly, splenomegaly, hepatic steatosis, liver dysfunction and cardiomyopathy.ConclusionPatients with CGL are rare, genetic sequencing should conducted in children presented with generalized lack of adipose tissue. The metabolic conditions should be closely followed-up since genetic diagnosis. One of the mutations c.713G>A, p.Gly238Asp in theBSCL2 gene of this patient is a novel mutation, which has not been reported so far.
Congenital generalized lipodystrophy; seipin; Genetic Sequencing; Hyperlipidemia; Acanthosis nigricans
1 福建省福州儿童医院内分泌科,福建医科大学教学医院 福州,350005;2 上海交通大学医学院附属上海儿童医学中心 上海,200127
陈瑞敏,E-mail: chenrm321@sina.com
10.3969/j.issn.1673-5501.2016.05.013
2016-07-25
2016-10-10)
