GFM1突变所致儿童急性肝衰竭1例:质疑GFM1错义突变位置决定临床表型
2016-11-26尤艺杰Agntig王建设
尤艺杰 Agnès Rötig 王建设
·论著·
GFM1突变所致儿童急性肝衰竭1例:质疑GFM1错义突变位置决定临床表型
尤艺杰1Agnès Rötig2王建设
目的 报告GFM1突变所致疾病的临床特征及验证基因型和临床表型关系。方法 回顾性分析1例门诊就诊的GFM1基因突变儿童的临床及基因突变资料,结合文献归纳GFM1基因突变的临床特点,构建编码线粒体翻译因子G1(mtEFG1)空间结构图,检验GFM1基因错义突变位置与临床表型关系的假说。结果 患儿,女,6个月28 d。生长发育迟缓,急性肝衰竭。体格检查:反应差,嗜睡,全身皮肤黏膜黄染,腹平软,肝脾肿大。辅助检查:总胆红素、直接胆红素、转氨酶、碱性磷酸酶、血清γ-谷氨酰转肽酶和血氨升高,白蛋白下降,凝血酶原时间延长,血糖下降,酸中毒,血乳酸升高,血浆氨基酸及酰基肉碱谱示血中多种氨基酸升高,尿有机酸检查提示酮尿,头颅MRI示双侧丘脑、大脑脚、基底节区、枕叶前内侧异常信号。GFM1基因的复合杂合突变,第5外显子c.688G>A点突变致蛋白质改变为p.Gly230Ser;第14外显子c.1686delG的移码突变致蛋白质改变为p.Asp563Thrfs*24。mtEFG1空间结构图显示,p.Gly230Ser处于蛋白周边位置,预测表现应该为脑型。结论 1例携带GFM1基因复合杂合突变(c.688G>A和c.1686delG)的中国儿童,其临床表现为急性肝衰竭,不支持以往GFM1基因错义突变导致蛋白周边位置的氨基酸改变时临床表现为脑型的假设。
GFM1; 线粒体翻译因子G1; 复合氧化呼吸链缺陷; 线粒体翻译因子G1; 肝衰竭
1 病例报告
患儿,女,6月28 d,因“皮肤巩膜黄染6月余”来复旦大学附属金山医院儿科(我院)就诊。患儿,G2P2,孕35+1周,因羊水偏少行剖腹产,出生体重1 700 g。患儿哥哥,40+1周剖腹产,出生体重 2 450 g,生后12 h内出现酸中毒,1周龄死于呼吸衰竭。母亲2次孕期均有皮肤瘙痒,第2次孕期血总胆汁酸28.9 μmol·L-1(参考值0~13 μmol·L-1)生后1 d因“气促”入当地医院,查CK和CK-MB升高,肝肾功能正常,头颅CT示早产儿未成熟影像,X线胸片示双肺纹理增强、模糊,抗感染治疗13 d后好转出院。1个月13 d患儿因“全身皮肤黄染1月,面色苍白15 d”再入当地医院,Hb 104 g·L-1,总胆红素(TB) 117.5 μmol·L-1,直接胆红素(DB) 53.6 μmol·L-1,乳酸(Lac)>20 mmol·L-1,凝血酶原时间(PT) 34.7 s(表1),头颅彩超示脑室内出血,头颅MRI示双侧脑白质重度髓鞘化障碍、脑皮层部分软化,尿有机酸检测:乳酸、2-羟基丁酸、丙酮酸、3-羟基丁酸升高,血浆氨基酸及酰基肉碱谱:丙氨酸、甲硫氨酸、苯丙氨酸、酪氨酸和脯氨酸升高,输注新鲜冰冻血浆、输血以及碳酸氢钠纠酸治疗28 d,患儿持续表现为凝血障碍、酸中毒和高乳酸血症,家属自动要求出院,出院时仍有全身皮肤轻度黄染。4个月28 d患儿因“咳嗽、痰鸣2 d加重伴气促1 d” 第3次入当地医院,血糖2.31 mmol·L-1,TB 259.7 μmol·L-1,PT 28.8 s,Lac 18.85 mmol·L-1(表1),双肺闻及明显痰鸣音,X线胸片示双肺纹理增强;头颅MRI:双侧丘脑、大脑脚、基底节区、枕叶前内侧异常信号,双侧额颞顶叶倾向于软化灶。予维生素K1预防出血,碳酸氢钠纠酸,复合辅酶和促肝细胞生长素护肝,抗感染。住院10 d后病情加重,家属自动要求出院。

表1 患儿发病期间的实验室检查指标
注 TB:总胆红素( μmol·L-1);DB:直接胆红素( μmol·L-1);AST和ALT(U·L-1);TBA: 总胆汁酸(μmol·L-1); GGT:γ谷氨酰转肽酶( U·L-1);CK-MB(U·L-1);BE:剩余碱(mmol·L-1); Lac:乳酸( mmol·L-1);PT:凝血酶原时间(s);INR:国际标准化比值
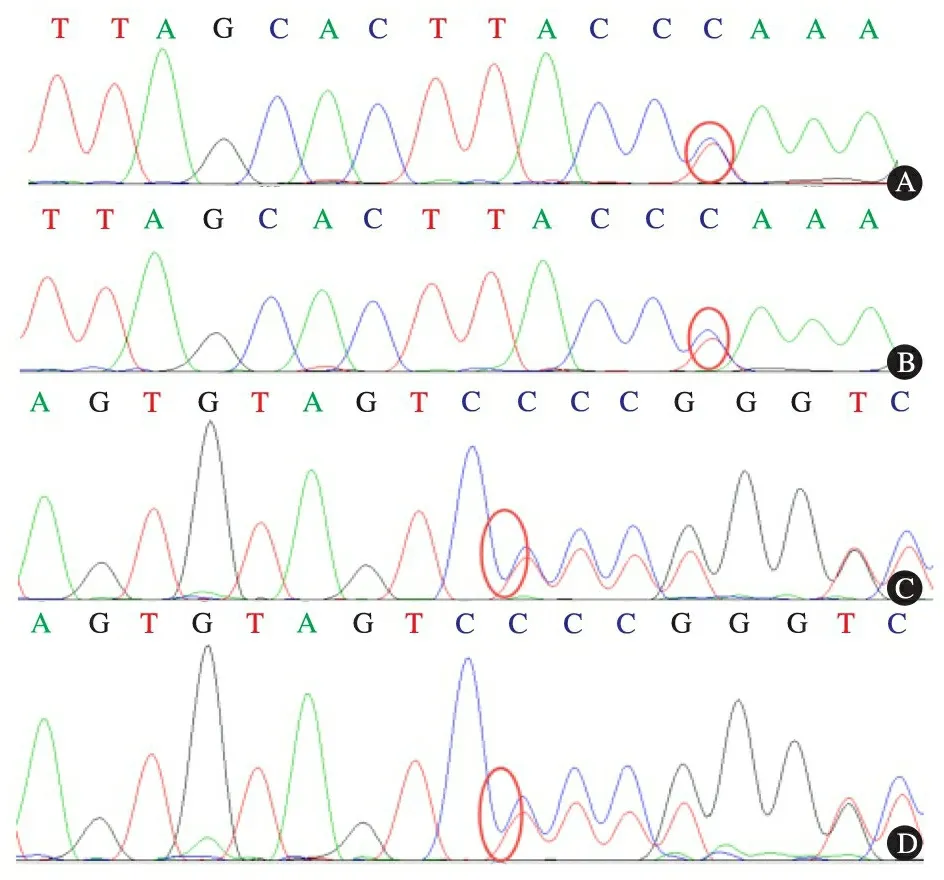
患儿来我院就诊,体格检查发现生长发育迟缓(体重为4.2 kg,WHO儿童生长曲线 在知情同意后抽取患儿及其父母外周血各2 mL(EDTA抗凝),提取DNA(TIANGEN试剂盒提取患儿及父母外周血DNA),患儿行代谢性肝病panel检测(北京迈基诺基因科技有限责任公司)。由UCSC网站下载GFM1基因组全序列(参考序列: NM_024996),获取GFM1基因相关信息,并用Primer 3设计PCR引物,双向测序法进行基因序列分析。患儿存在GFM1基因的复合杂合突变,第5个外显子c.688G>A突变(图1A)和第14个外显子c.1686delG突变(图1C),突变分别来自父亲(图1B)和母亲(图1D)。c.688G>A预测引起p.Gly230Ser改变,为已知致病性突变[1],c.1686delG引起框移后造成终止密码提前出现。结合患儿起病年龄和临床表现,诊断为GFM1基因突变引起的肝功能衰竭。患儿回当地等待基因检测结果中死亡。 复习文献,自Galmiche提出错义突变的空间位置决定基因型和表型关系的假设以来,又新发现了2种错义突变(K304E和G230S)[1,2]。本研究运用Galmiche报道的方法,构建了线粒体翻译因子G1(mtEFG1)蛋白模型三维空间(图2)。发现K304E和G230S均位于mtEFG1蛋白周边位置。K304E的临床表型为脑型,符合上述假设;而G230S的临床表型为肝型,并不符合上述假设。 图1 患儿及父母基因测序图 注GFM1基因的复合杂合突变,第5外显子c.688G>A的点突变(A),突变来自父亲(B);第14外显子c.1686delG的移码突变(C),突变来自母亲(D);红色圆圈示突变位点 图2 8种错义突变在mtEFG1蛋白模型三维空间的位置 注 红色字体临床表现为肝型;黄色字体临床表现为脑型 GFM1基因突变所致疾病为罕见的累及多系统的常染色体隐性遗传病,可引起复合氧化磷酸化呼吸链(OXPHS)缺陷[3], Coenen 等在2004年首次报道,目前全球只有12例报道,10例患儿在出生后不久即发病(6例于出生后1周内发病,4例出生后2个月内发病)[1~8],进展迅速,其中8例患儿在4岁前死亡[1,3~8],1例患儿死亡年龄未知[2],只有1例患儿在5岁6个月依然存活[7]。Balasubramaniam等[1]在2012年曾报道1中国裔儿童,为c.688G>A和c.539delG复合杂合突变,临床表现为肝型[1]。本文报告病例为第2例中国儿童GFM1突变病例报道,为c.688G>A和c.1686delG复合杂合突变。2例中国儿童GFM1突变病例都有一个共同位点的错义突变,该突变在11例非中国儿童中未见报道,推测该突变位点可能为中国儿童的热点突变。 GFM1基因位于3q25.1~q26.2,共48 044 bp,18个外显子,mtEFG1[9]在mtDNA翻译过程中发挥作用[10]。致mtEFG1改变的GFM1基因突变阻碍了mtDNA编码13种 OXPHOS 蛋白亚基(这13 种OXPHOS 蛋白亚基分别是复合体Ⅰ、Ⅲ、Ⅳ、Ⅴ的重要组成部分),表现为复合体I、或(和)Ⅲ、或(和)Ⅳ、或(和)Ⅴ的活性下降,复合氧化磷酸链缺陷,电子传递过程或ADP磷酸化障碍,ATP生成减少,能量代谢障碍,器官功能受限[10~12]。 Valente等在2007年首次提出mtEFG1蛋白突变位置与临床表现有关的假设[8]。Galmiche等在2012年对当时已报道的6种错义突变进行分析,发现突变发生在mtEFG1蛋白中间位置(N174S,S321P,L398P)时临床表现为肝脏表型,而突变发生在此蛋白周边位置(M496R,R250W,R671C)表现为脑型[6],并因此提出假设:GFM1基因错义突变发生在蛋白中间位置时,临床表现为肝型;相反,则临床表现为脑型[6,7]。本文分析了继Galmiche等文章后又发现的2种新错义突变,发现一种符合上述假说,一种反对上述假说,说明错义突变的氨基酸位置决定临床表现型需要完善,或者是错误的。 GFM1基因突变引起的复合氧化呼吸链缺陷病可累及多个系统,临床表现多样,目前主要根据GFM1基因测序进行诊断,无特异性治疗,预后极差[13~16]。GFM1基因突变基因型和临床表现型的关系尚需要进一步研究。 [1]Balasubramaniam S, Choy YS, Talib A, et al. Infantile Progressive Hepatoencephalomyopathy with Combined OXPHOS Deficiency due to Mutations in the Mitochondrial Translation Elongation Factor Gene GFM1. JIMD Rep, 2012, 5:113-122 [2]Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med, 2012, 4(118):118ra10 [3]Coenen MJ, Antonicka H, Ugalde C, et al. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med, 2004, 351(20):2080-2086 [4]Smits P, Antonicka H, van Hasselt PM, et al. Mutation in subdomain G' of mitochondrial elongation factor G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur J Hum Genet, 2011, 19(3):275-279 [5]Antonicka H, Sasarman F, Kennaway NG, et al. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet, 2006, 15(11):1835-1846 [6]Galmiche L, Serre V, Beinat M, et al. Toward genotype phenotype correlations in GFM1 mutations. Mitochondrion, 2012, 12(2): 242-247 [7]Brito S, Thompson K, Campistol J, et al. Long-term survival in a child with severe encephalopathy, multiple respiratory chain deficiency and GFM1 mutations. Front Genet, 2015, 6: 102 [8]Valente L, Tiranti V, Marsano RM, et al. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet, 2007, 80(1):44-58 [9]Hammarsund M, Wilson W, Corcoran M, et al. Identification and characterization of two novel human mitochondrial elongation factor genes, hEFG2 and hEFG1, phylogenetically conserved through evolution. Hum Genet, 2001, 109(5):542-550 [10]Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol, 2010: 737385 [11]van Waveren C, Moraes CT. Transcriptional co-expression and co-regulation of genes coding for components of the oxidative phosphorylation system. BMC Genomics, 2008, 9:18 [12]Saada A. Mitochondria: mitochondrial OXPHOS (dys) function ex vivo--the use of primary fibroblasts. Int J Biochem Cell Biol, 2014, 48:60-65 [13]Soiferman D, Ayalon O, Weissman S, et al. The effect of small molecules on nuclear-encoded translation diseases. Biochimie, 2014, 100: 184-191 [14]Tonin Y, Heckel AM, Dovydenko I, et al. Characterization of chemically modified oligonucleotides targeting a pathogenic mutation in human mitochondrial DNA. Biochimie, 2014, 100:192-199 [15]Ribas GS, Vargas CR, Wajner M. L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene, 2014, 533(2):469-476 [16]Romano S, Samara D, Crosnier H, et al. Variable outcome of growth hormone administration in respiratory chain deficiency. Mol Genet Metab, 2008, 93(2):195-199 (本文编辑:张崇凡) Acute liver failure caused by GFM1 mutations in a child: the relationship between GFM1 missense mutation and the peripheral amino acid and the change of clinical phenotype YOU Yi-jie1, Agnès Rötig2, WANG Jian-she3 (1 Department of pediatrics, Jinshan Hospital of Fudan University, Shanghai 201508, China;2 Department of Genetics, Hpital Necker-Enfants Malades, Université Paris Descartes and INSERM U781, 149 rue de Sèvres,75015 Paris, France;3 Department of infectious diseases, Children Hospital of Fudan University, Shanghai 201102, China) WANG Jian-she,E-mail: jshwang@shmu.edu.cn Objective To summarize the clinical characteristics in children harboringGFM1 gene mutations.MethodsRetrospective analysis about the clinical features was performed in a patient with biallelicGFM1 mutations. Mitochondrial translation factor G1 (mtEFG1) space structure was constructed to verify the hypothesis about the relation betweenGFM1 gene missense mutations and the results of clinical phenotypes.ResultsThe patient was a girl aged six months and twenty-eight days. Growth retardation, abnormal liver function, jaundice, hepatomegaly and splenomegaly were presented with in poor response and lethargy. Serum biochemical tests demonstrated elevation in total bilirubin, direct bilirubin, aspartate transaminase, alkaline phosphatase, gamma-glutamyltransferase, blood ammonia and blood lactic acid, as well as hypoalbumineamia, prolonged PT, acidosis, and hypoglycemia.Mass spectrometry of serum amino acids & acyl carnitine and urine urine organic acids suggested elevation of many amino acids in serum and ketonuria. Abnormal signals were observed in bilateral thalamus, cerebral peduncle, basal ganglia, and medial anterior occipital lobes through cranial MRI. Sequencing ofGFM1 revealed compound heterozygous mutations: a missense mutation c.688G>A (p.Gly230Ser) in exon 5 and a frameshift deletion c.1686delG (p.Asp563Thrfs*24) in exon 14. Gly230Ser protein changed amino acid residue in the structure of peripheral mtEFG1 space, changes in which area resulted in encephalopathy.ConclusionA Chinese girl carrying compound heterozygousGFM1 gene mutations of c.688G>A and c.1686delG was reported. Hepatic presentation of this case denied the assumption that missense mutations inGFM1 gene that changes the peripheral amino acids of the protein will cause phenotype of encephalopathy. GFM1; MtEFG1; Composite oxide respiratory chain defects; MtDNA; Liver failure 国家自然科学基金资助项目:81570468 1 复旦大学附属金山医院儿科 上海,201508;2 Department of Genetics, Hpital Necker-Enfants Malades, Université Paris Descartes and INSERM U781, 149 rue de Sèvres,Paris, France;3 复旦大学附属儿科医院感染科 上海,201102 王建设 ,E-mail: jshwang@shmu.edu.cn 10.3969/j.issn.1673-5501.2016.05.011 2016-05-30 2016-09-28)2 基因型和表型关系分析

3 讨论

