添加氯离子对Ni/CeO2催化剂CO选择甲烷化反应催化性能的促进
2016-11-25高志明代倩子马宏伟
高志明, 代倩子, 马宏伟
(北京理工大学 化学学院,北京 102488)
添加氯离子对Ni/CeO2催化剂CO选择甲烷化反应催化性能的促进
高志明, 代倩子, 马宏伟
(北京理工大学 化学学院,北京 102488)
采用浸渍方法制备NiO(Clx)/CeO2系列样品,经还原后得到Ni(Clx)/CeO2催化剂,用于富氢气体中CO的选择甲烷化反应,考察Cl/Ce原子比(x)对CO选择甲烷化反应活性的影响. 结果表明,添加氯离子能够显著抑制CO2甲烷化反应,因而可使富氢气体中CO浓度降至10-5以下并且CO甲烷化反应选择性不低于50%,满足质子交换膜燃料电池对燃料的要求. 通过CO和CO2吸附实验、程序升温还原(TPR)测定以及物相组成和晶粒尺寸分析,揭示添加氯离子的作用.
金属镍;二氧化铈;氯离子;CO选择甲烷化;氢气精制
质子交换膜燃料电池(PEMFC)可作为大型电站使用,也可作为小型发电装置使用,所用燃料系通过烃类物质的水蒸气重整反应、水气变换反应以及后续的氢气精制步骤制备得到. 重整-变换反应后的富氢气体的大致组成为:0.5%~2.0% CO、15%~20% CO2、约70% H2和10%水蒸气(体积分数). 此浓度的CO将导致PEMFC的电极中毒,须将其降至10-4以下(对应Pt-Ru电极)甚至10-5以下(对应Pt电极)[1]. 对于PEMFC应用于大型电站的场合,可利用变压吸附(PSA)装置进行氢气精制;而对于小型发电装置的场合,由于空间的限制,只能通过催化反应的方法除去CO. 目前国外市场销售的家庭用小型PEMFC装置,系采用CO优先氧化反应(CO-PROX)使CO转化成CO2,但是同时也伴随有一定量H2被氧化生成H2O[2]. 所用催化剂为贵金属负载催化剂. 另外,该方法还需要外加空气以实现氧化反应,导致装置复杂化.
近年来,富氢气体中CO选择甲烷化反应(CO-SMET)受到重视,因为该反应不需要外加反应气体,同时还可使用价格低廉的金属镍催化剂[1-6]. CO-SMET反应催化剂开发的关键是,既要对CO甲烷化反应具有高活性,同时又要抑制CO2甲烷化反应以减少氢气消耗. CO甲烷化反应选择性可定义为,CO甲烷化所需氢气量与CO和CO2甲烷化所需总氢气量之比. CO-SMET反应的优良催化剂,须能够使CO浓度降至10-4甚至10-5以下,同时CO甲烷化反应选择性不低于50%.
CO-SMET反应催化剂的常用载体有TiO2、Al2O3、CeO2、ZrO2等[5]、活性组分有金属Ru和Ni[6]. 载体种类影响催化性能. 文献[7]报道,在负载金属Ru催化剂上,CO转化率和CO2转化率按大小顺序,按载体种类排列为TiO2,Al2O3,CeO2,YSZ,SiO2. 金属Ru的催化活性优于金属Ni[6]. 向Ni/Al2O3催化剂中添加少量金属Ru可提高CO-SMET反应催化性能,使CO浓度降至10-4以下[4]. Ni/CeO2催化剂被用于CO-SMET反应,由于CO2转化率高而导致了CO甲烷化反应选择性过低;添加氯离子极大地抑制了CO2甲烷化反应[8]. 许多文献认为氯离子能够抑制CO和CO2吸附[8-9],但是缺少吸附量测定数据. 关于添加氯离子对物相组成和晶粒尺寸的影响,文献上也缺少实验数据. 为了进一步系统阐明添加氯离子的作用,本文工作制备了Ni(Clx)/CeO2系列催化剂,其中Cl/Ce原子比x=0~0.30,测定有无氯离子添加情况下的CO和CO2吸附量,考察氯离子对物相组成和晶粒尺寸以及样品还原性质的影响.
1 实 验
1.1 催化剂制备
称取适量硝酸铈(Ce(NO3)3·6H2O,A.R.,国药集团),放入马弗炉中,在500 ℃下热分解1 h,得到二氧化铈粉末,研磨后作为载体使用.
采用浸渍方法制备催化剂样品. 首先,称取适量镍盐(Ni(NO3)2·6H2O,A.R.,北京通广精细化工公司. NiCl2·6H2O,A.R.,国药集团),溶于去离子水中. 其中氯化镍与硝酸镍的相对质量系根据投料Cl/Ce原子比确定. 在无氯离子添加的情况下,仅称取硝酸镍. 将适量二氧化铈粉末加入到镍盐水溶液中,充分搅拌后,转移入旋转蒸发器中,在80 ℃、真空度0.08 MPa下蒸发除去多余水分. 然后,在烘箱中80 ℃下干燥5 h,继续在110 ℃下干燥14 h. 将干燥后的样品放入马弗炉中,在500 ℃下焙烧2 h,得到氧化态样品mNiO(Clx)/CeO2. 其中,m为投料Ni/Ce原子比(以百分数表示);x为投料Cl/Ce原子比(以小数表示).
在CO-SMET反应前,将氧化态样品在20%H2/N2混合气中500 ℃下还原2 h,得到还原态样品即催化剂mNi(Clx)/CeO2.
1.2 催化剂表征
样品中实际Ni/Ce原子比由诱导耦合等离子体原子发射光谱(ICP-AES)测定(Thermo Scientific, iCAP 6000 Series).
对氧化态样品和反应后催化剂样品进行X射线衍射(XRD)分析(D8 Advance, Bruker),Cu Kα射线,管电压40 kV,管电流40 mA. 利用Rietveld方法计算样品的物相组成和CeO2晶粒尺寸. 对含量低的物相NiO或金属Ni,利用谢乐方程计算特定晶面方向上的厚度.
利用程序升温还原(TPR)方法测定氧化态样品的还原性质(PX200,天津市鹏翔科技). 10%H2/Ar混合气流速40 mL/min,升温速率10 ℃/min,样品质量50.0 mg. 重复测定两次,取TPR峰面积平均值,相对误差在5%以内. 仪器响应因子由NiO标准样品确定.
对氧化态样品进行比表面积(SSA)测定(JW-DA,北京精微高博). 样品首先在150 ℃下真空脱气处理1 h,然后在液氮温度(-196 ℃)下吸附氮气,相对压力p/p0=0.06 ~ 0.30,利用BET方程计算比表面积.
氧化态样品和反应后催化剂样品的表面元素组成由X光电子能谱(XPS)分析测定(PHI, Quantera),Al Kα射线,分析腔压力为10-8Pa. 结合能数值由污染碳C1s=284.8 eV进行校正.
1.3 催化活性评价
催化剂活性评价在常压连续流动固定床反应装置上进行. 称取200 mg氧化态样品颗粒(60~80目),装入石英反应管(内径8 mm)中,样品两端由石英棉固定. 控温热电偶位于石英管内石英棉层内紧邻样品. 首先,对氧化态样品进行还原处理. 20%H2/N2还原气流速100 mL/min,在500 ℃下还原2 h,然后在还原气中降至室温. 切换成反应气体,组成为1%CO,18%CO2,70%H2,11%N2(体积分数),流速50 mL/min. 催化反应采用升温方式进行. 在任一反应温度下稳定1 h后通过六通阀取样,在气相色谱仪(GC9800,上海科创)上进行产物组成分析. 热导池检测器(TCD),氦气作载气,填充柱为Shincarbon ST(日本岛津),可分离N2、CO、CH4、CO2、C2烃等. 以N2作内标,计算CO转化率(XCO)、CO2转化率(XCO2)和CO甲烷化反应选择性(S),如式(1) ~ (3)所示. CO和CO2的加氢产物为甲烷,没有检测到其它烃生成. 本文所报道催化活性数据皆为两次实验的平均值,相对误差一般在5%以内.
(1)
(2)
(3)
式中:ACO_0、AN2_0和ACO2_0分别为反应前气体中CO、N2和CO2的色谱峰面积;ACO_1、AN2_1和ACO2_1分别为反应后气体中CO、N2和CO2的色谱峰面积.
催化反应结束后,将催化剂样品在反应气中降至室温. 然后,切换至1%O2/He混合气(50 mL/min)钝化0.5 h. 对反应后催化剂样品(钝化状态)进行XRD和XPS测试.
1.4 CO和CO2吸附量测定
在智能重量分析仪(IGA100C, Hiden Isochema Ltd, UK)上测定CO和CO2吸附等温线. 称取70 mg氧化态样品粉末,放入样品篮中,置于分析仪内. 系统密封后,抽至高真空. 然后,通入纯氢气至20 kPa并保持氢气流速60 mL/min,同时以3 ℃/min升至500 ℃、恒温0.5 h. 在该氢气气氛中降至室温,然后再次抽至高真空. 向系统中通入纯CO(或CO2)气体,设定压力上升速率3.3 kPa/min,终止压力100 kPa. 记录CO(或CO2)气体压力上升过程中样品质量的变化,得到室温下CO(或CO2)的吸附等温线.
2 结果与讨论
图1是氧化态样品20%NiO(Clx)/CeO2(x=0~0.30)的XRD图谱. 对所有样品均可见微弱的NiO物相的衍射峰(PDF 71-1179),对应2θ=37.2°,43.3°,62.9°. 其它衍射峰归属于CeO2物相(PDF 89—8436).
利用Rietveld方法对图1中诸样品的XRD衍射峰进行拟合. 结果列于表1中.
表1 样品20%NiO(Clx)/CeO2(x=0~0.30)中NiO质量分数及各相晶粒尺寸
Tab.1 Weight percents of NiO and crystallite sizes of samples 20%NiO(Clx)/CeO2(x=0~0.30)
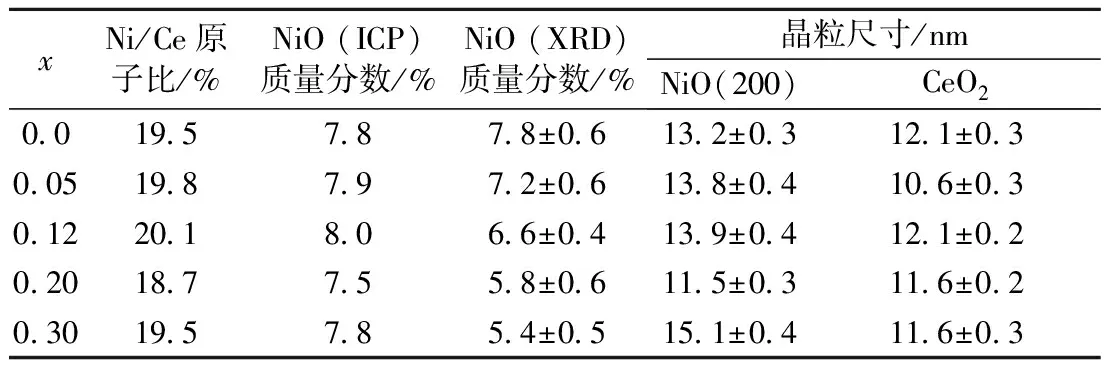
xNi/Ce原子比/%NiO(ICP)质量分数/%NiO(XRD)质量分数/%晶粒尺寸/nmNiO(200)CeO2001957878±06132±03121±030051987972±06138±04106±030122018066±04139±04121±020201877558±06115±03116±020301957854±05151±04116±03
注:Ni/Ce原子比为ICP测定值;NiO(ICP)和NiO(XRD)为分别由ICP测试结果和Rietveld方法得到的样品中NiO质量分数;NiO(200)为由谢乐方程计算得到的NiO晶粒在(200)晶面上的厚度.
可见,各样品中CeO2物相的晶粒尺寸相近,在11.6 nm左右. NiO物相衍射峰较弱,故利用谢乐方程计算了样品中NiO晶粒在垂直于(200)晶面(2θ=43.3°)方向上的厚度,结果表明各样品中NiO晶粒也具有相似的尺寸,在13.5 nm左右. 利用X光电子能谱(XPS)测定了催化反应前后的投料Cl/Ce原子比x=0,0.20样品的表面元素组成.
如表2所示,对于x=0.20的样品,在催化反应前后,表面Cl/(Ce+Ni)原子比很相近,表明氧化态样品20%NiO(Cl0.2)/CeO2在500 ℃下20%H2/N2混合气中还原2 h未导致氯离子明显流失,因而能够保持样品表面Cl/(Ce+Ni)原子比在催化反应前后基本保持不变. 这与文献[8]结果一致. 在样品中氯离子与金属离子处于键合状态. 由于样品系通过CeO2浸渍镍盐溶液的方法制备得到,故在氧化态样品中Cl-将主要与Ni2+处于键合状态. 因而,随着氧化态样品中氯离子含量增加,形成NiO晶粒的Ni2+数量将减少. 这与表1中的Rietveld方法拟合结果一致,即随着投料Cl/Ce原子比增加,NiO物相组成百分数有所下降. 与许多文献[8,10-12]类似,本文也未测定样品中氯离子的实际含量. 氯离子含量可通过离子色谱测定,但是离子色谱尚不是普及型分析仪器. 本文所采用焙烧条件为马弗炉中500 ℃下2 h. 即使在焙烧过程中氯离子有部分损失,也可以认为,投料Cl/Ce原子比越高,焙烧后样品中剩余氯离子量越大. 文献[11]指出,在500 ℃以上焙烧时才发生氯离子的明显流失.
表2 样品20% NiO(Clx)/CeO2(记为f)及反应后催化剂20% Ni(Clx)/CeO2(记为s) (x=0, 0.20)的表面元素组成
Tab.2 Surface element composition of the fresh samples 20%NiO(Clx)/CeO2(denoted asf) and the spent catalysts 20%Ni(Clx)/CeO2(denoted as s) (x=0, 0.20)
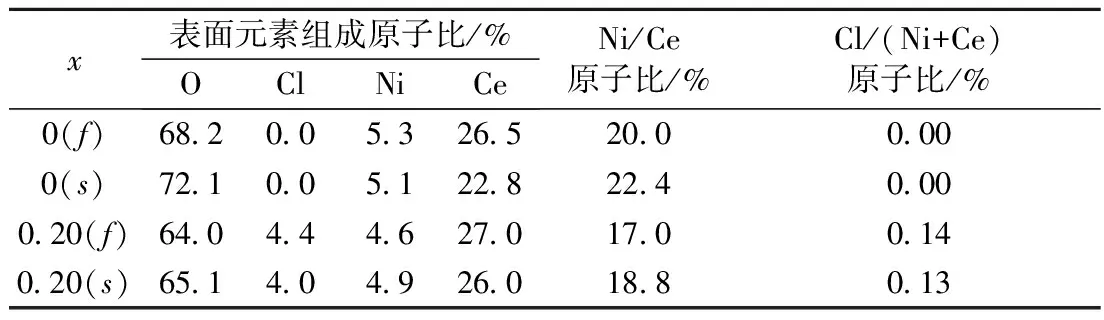
x表面元素组成原子比/%OClNiCeNi/Ce原子比/%Cl/(Ni+Ce)原子比/%0(f)68200532652000000(s)7210051228224000020(f)6404446270170014020(s)6514049260188013
图2是氧化态样品20%NiO(Clx)/CeO2(x=0~0.30)的TPR曲线. 可见,在无氯离子添加时,出现3个还原峰,表明有不同状态的Ni2+存在[13]. 随着添加氯离子量增加,逐渐形成1个还原峰并且峰位向高温移动. 这表明氯离子有使Ni2+状态均一化的作用并且使Ni2+还原性降低. 类似地,CuO/CeO2样品也出现多重TPR峰[14-15],其被认为源于不同状态的Cu2+,即与CeO2有强相互作用的CuO、在载体上良好分散的CuO簇以及CuO晶粒等[14-16]. 对图2中各样品的TPR峰面积进行了积分. 结合ICP测得的镍含量及TPR仪器的响应因子,发现每个样品的TPR峰面积均大于全部Ni2+还原成金属镍所对应的峰面积,表明载体CeO2发生了伴生还原(形成CeO2-d). 这与文献报道一致[17]. 对CuO/CeO2样品,许多文献[14,18]也报道在TPR过程中发生载体CeO2的伴生还原. 对样品NiO(Clx)/CeO2,有文献认为载体CeO2发生部分还原后可形成CeOCl化合物[8].
表3列出了样品20%NiO(Clx)/CeO2(x=0~0.30)中载体CeO2在TPR过程中发生的伴生还原程度(即CeO2-d的d值). 可见,添加氯离子导致载体CeO2伴生还原程度降低,但是在x=0.12~0.30范围内d值恒定在0.13左右.d值的误差约为±10%. 氯离子能够抑制负载金属对氢的吸附,因而将导致从金属到载体的氢溢流减少[19-20]. 另外,还可看到,无氯离子添加时样品的比表面积为68 m2/g,当x增加到0.12~0.30时,样品的比表面积下降至58 m2/g左右. 这与文献[8]结果一致.
表3 样品20% NiO(Clx)/CeO2(x=0~0.30)的比表面积和TPR过程中载体的伴生还原程度
Tab.3 Thedvalue in CeO2-dformed in TPR process and specific surface area (SSA) of samples 20% NiO(Clx)/CeO2(x=0~0.30)
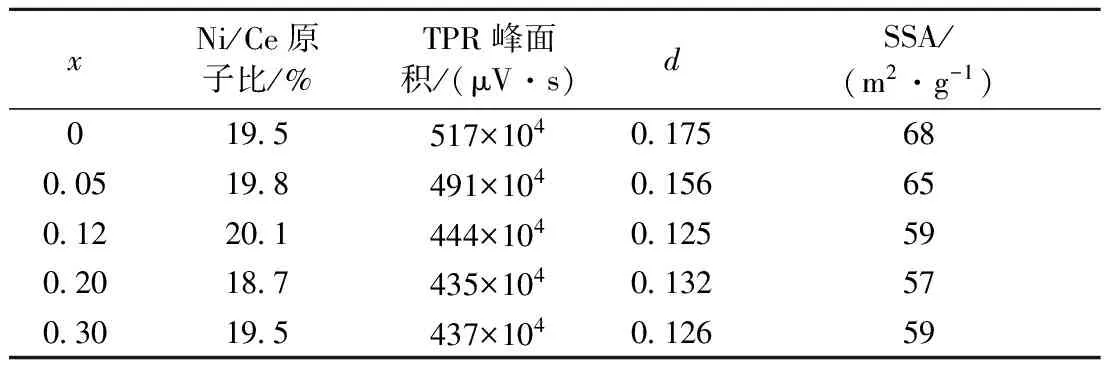
xNi/Ce原子比/%TPR峰面积/(μV·s)dSSA/(m2·g-1)0195517×104017568005198491×104015665012201444×104012559020187435×104013257030195437×104012659
注:Ni/Ce原子比为ICP测定值.
对氧化态样品20%NiO(Clx)/CeO2在20%H2/N2混合气中500 ℃下还原2 h,得到催化剂20%Ni(Clx)/CeO2,进行了催化性能评价. 结果示于图3中. 可见,添加氯离子能够显著抑制CO2转化率;并且,由于氯离子对CO2转化的抑制作用,使得CO转化率得到提高. 在无氯离子添加(即x=0)时,CO转化率在反应温度220 ℃时可达100%,但是在230 ℃时便降至99.2%,随后在270 ℃时继续降至98.9%. 一般认为,CO2甲烷化反应由两步构成[21-23]:第1步是逆水气变换反应,导致CO生成;第2步是CO加氢生成甲烷的反应. 所以,当CO2转化率升高时,若CO2没有被完全加氢至甲烷而是有中间产物CO生成,则将导致CO表观转化率下降. 在x=0.20时,CO转化率在反应温度210 ℃达100%,并一直保持到270 ℃. 但是,当x=0.30时,CO转化率在210 ℃时为98.9%,说明氯离子过量添加又抑制了CO转化率. 所以,本文的氯离子最佳添加量为x=0.20. 文献[8]中也报道存在最佳氯离子添加量,其最佳氯离子添加量为x=0.12.
反应后催化剂样品20%Ni(Clx)/CeO2的XRD谱图示于图4中. 金属Ni晶粒的衍射峰在2θ=44.3°和51.7°(PDF 89—7128)处微弱可见. Rietveld方法拟合计算表明,各催化剂样品中金属Ni晶粒含量相近,如表4所示,在2.3%(质量分数)左右. 这明显低于投料质量分数,表明一部分金属Ni高度分散在载体上、未被XRD检测到. 由于金属Ni晶粒含量低,故利用谢乐方程计算了Ni晶粒在垂直于(111)晶面(2θ=44.3°)方向上的厚度. 由表4可见,随着投料Cl/Ce原子比增加,Ni晶粒尺寸呈现增加的趋势. 另外,由于反应后催化剂样品经过1%O2/N2混合气钝化,故本文不对催化剂样品中铈离子价态进行讨论.
表4 催化剂20%Ni(Clx)/CeO2(x=0~0.30)中金属Ni质量分数及各相晶粒尺寸
Tab.4 Weight percents of metal Ni and crystallite sizes of catalysts 20%Ni(Clx)/CeO2(x=0~0.30)
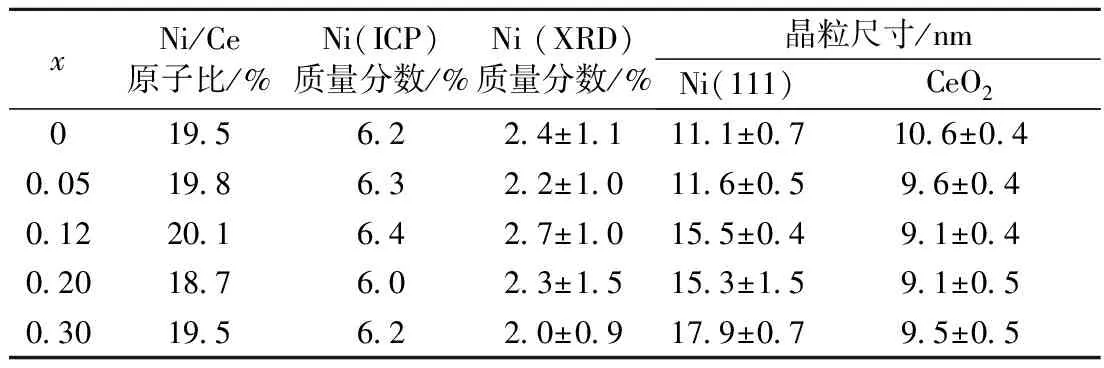
xNi/Ce原子比/%Ni(ICP)质量分数/%Ni(XRD)质量分数/%晶粒尺寸/nmNi(111)CeO201956224±11111±07106±040051986322±10116±0596±040122016427±10155±0491±040201876023±15153±1591±050301956220±09179±0795±05
注:Ni/Ce原子比为ICP测定值;Ni(ICP)和Ni(XRD)为分别由ICP测试结果和Rietveld方法得到的样品中金属Ni质量分数;Ni(111)为由谢乐方程计算得到的金属Ni晶粒在(111)晶面上的厚度.
许多文献认为氯离子能够抑制CO和CO2的吸附[8-9,10,19],但是缺少吸附量测定数据. 本文对还原态样品20%Ni(Clx)/CeO2(x=0, 0.20)测定了CO和CO2吸附等温线,结果如图5所示. 可见,添加氯离子极大地抑制了CO和CO2吸附量;在x=0.20时CO2吸附量被明显降低. 因而添加氯离子能够显著抑制CO2转化率(见图5).
本文工作针对系列催化剂样品mNi(Cl0.2)/CeO2(m=10%,20%,30%,40%),考察了金属Ni负载量的影响. 发现CO转化率和CO2转化率都随金属Ni负载量增加而增加. 这与文献结果一致[8]. 在m=20%时,在本文所选反应温度范围内CO2转化率较低. 故本文选择m=20%考察了添加氯离子的影响以及20%Ni(Cl0.2)/CeO2催化剂的催化性能稳定性. 在催化性能稳定性评价实验中,采用氢火焰检测器(FID)检测反应管出口CO和CH4浓度. 其结果示于图6中. 可见,在反应温度210 ℃时,CO浓度为9×10-5. 这低于热导池检测器(TCD)的检测限,故在该反应温度下图3中给出的CO转化率为100%. 图6表明,在反应温度220~250 ℃范围内CO浓度均低于10-5,在220~240 ℃范围内CO甲烷化反应选择性高于50%,在250 ℃时CO甲烷化反应选择性为43%. 随着反应温度升高,CH4浓度增加. 这是由于CO2转化率随反应温度升高而增加的结果.
3 结 论
对氧化态样品20%NiO(Clx)/CeO2(x=0~0.30),无论氯离子添加与否,在TPR过程中载体CeO2都发生伴生还原(生成CeO2-d). 其还原度(即d值)随着氯离子添加而减小,但是在x=0.12 ~ 0.30时d值基本恒定在0.13. 氯离子添加使样品中Ni2+状态趋于均一化;并且氯离子添加量越大,Ni2+趋于越难还原. 氯离子添加量影响催化剂20%Ni(Clx)/CeO2中金属Ni晶粒的尺寸. 可以预期,氯离子也将影响催化剂中金属Ni的电子状态. 在氧化态样品中氯离子主要以与Ni2+键合的状态存在,在还原态样品(催化剂)中认为氯离子可以CeOCl结构形式存在.
添加氯离子显著抑制CO和CO2吸附量,最终表现为极大地降低了CO2转化率、促进CO的深度除去. 在催化剂20%Ni(Cl0.2)/CeO2上,在反应温度220~250 ℃范围内反应管出口CO浓度均低于10-5,在220~240 ℃范围内CO甲烷化反应选择性高于50%.
[1] Mohaideen K K, Kim W, Koo K Y, et al. Highly dispersed Ni particles on Ru/NiAl catalyst derived from layered double hydroxide for selective CO methanation[J]. Catalysis Communications, 2015,60:8-13.
[2] Tada S, Kikuchi R, Wada K, et al. Long-term durability of Ni/TiO2and Ru-Ni/TiO2catalysts for selective CO methanation [J]. Journal of Power Sources, 2014,264:59-66.
[3] Dai X, Liang J, Ma D, et al. Large-pore mesoporous RuNi-doped TiO2-Al2O3nanocomposites for highly efficient selective CO methanation in hydrogen-rich reformate gases [J]. Applied Catalysis B: Environmental, 2015,165:752-762.
[4] Aihosseinzadeh A, Nematollahi B, Rezaei M, et al. CO methanation over Ni catalysts supported on high surface area mesoporous nanocrystalline γ-Al2O3for CO removal in H2-rich stream [J]. International Journal of Hydrogen Energy, 2015,40(4):1809-1819.
[5] Takenaka S, Shimizu T, Otsuka K. Complete removal of carbon monoxide in hydrogen-rich gas stream through methanation over supported metal catalysts [J]. International Journal of Hydrogen Energy, 2004,29(10):1065-1073.
[6] Park D P, Lee D, Lee H C. Recent progress in selective CO removal in H2-rich stream [J]. Catalysis Today, 2009,139(4):280-290.
[7] Panagiotopoulou P, Kondarides D I, Verykios X E. Selective methanation of CO over Ru catalysts[J]. Applied Catalysis B: Environmental, 2009,88:470-478.
[8] Zyryanova M M, Snytnikov P V, Gulyaev R V, et al. Performance of Ni/CeO2catalysts for selective CO methanation in hydrogen-rich gas [J]. Chemical Engineering Journal, 2014,238:189-197.
[9] Miyao T, Shen W, Chen A, et al. Mechanistic study of the effect of chlorine on selective CO methanation over Ni alumina-based catalysts[J]. Applied Catalysis A: General, 2014,486:187-192.
[10] Djinovic P, Galletti C, Specchia S, et al. CO methanation over Ru-Al2O3catalysts: effects of chloride doping on reaction activity and selectivity[J]. Topics in Catalysis, 2011,54(16):1042-1053.
[11] Badri A, Binet C, Lavalley J-C. Surface-chlorinated ceria and chlorine-containing reduced Pd/CeO2catalysts, a FTIR study[J]. Journal of Physical Chemistry, 1996,100(20):8363-8368.[12] Leitenburg C, Trovarelli A, Kaspar J. A temperature-programmed and transient kinetic study of CO2activation and methanation over CeO2supported noble metals[J]. Journal of Catalysis, 1997,166(1):98-107.
[13] Malwadkar S, Bera P, Hegde M S, et al. Preferential oxidation of CO on Ni/CeO2catalysts in the presence of excess H2and CO2[J]. Reaction Kinetics Mechanisms and Catalysis, 2012,107(2):405-419.
[14] Caputo T, Lisi L, Pirone R, et al. On the role of redox properties of CuO/CeO2catalysts in the preferential oxidation of CO in H2-rich gases [J]. Applied Catalysis A: General, 2008,348:42-53.
[15] Tang X, Zhang B, Li Y, et al. CuO/CeO2catalysts: Redox features and catalytic behaviors [J]. Applied Catalysis A: General, 2005,288:116-125.
[16] Liu Y, Fu Q, Stephanopoulos M F. Preferential oxidation of CO in H2over CuO-CeO2catalysts [J]. Catalysis Today, 2004,93-95:241-246.
[17] Tada S, Shimizu T, Kameyama H, et al. Ni/CeO2catalysts with high CO2methanation activity and high CH4selectivity at low temperatures [J]. International Journal of Hydrogen Energy, 2012,37(7):5527-5531.
[18] Shan W, Shen W, Li C. Structural characteristics and redox behaviors of Ce1-xCuxOysolid solutions[J]. Chemistry of Materials, 2003,15(25):4761-4767.
[19] Kondarides D I, Verykios X E. Effect of chlorine on the chemisorptive properties of Rh/CeO2catalysts studied by XPS and temperature programmed desorption techniques[J]. Journal of Catalysis, 1998,174(1):52-64.
[20] Fajardie F, Tempere J F, Djega-Mariadassou G, et al. Benzene hydrogenation as a tool for the determination of the percentage of metal exposed on low loaded ceria supported rhodium catalysts[J]. Journal of Catalysis, 1996,163(1):77-86.
[21] Tada S, Kikuchi R, Takagaki A. Study of Ru-Ni/TiO2catalysts for selective CO methanation[J]. Applied Catalysis B: Environment, 2013,140-141:258-264.
[22] Urasaki K, Tanpo Y, Takahiro T, et al. Selective methanation of CO in reformate gas over Ni/TiO2catalyst[J]. Chemistry Letters, 2010,39:972-973.
[23] Shimoda N, Shoji D, Tani K, et al. Role of trace chlorine in Ni/TiO2catalyst for CO selective methanation in reformate gas[J]. Applied Catalysis B: Environment, 2015,174-175:486-495.
(责任编辑:李兵)
Effect of Chlorine Doped in Ni/CeO2Catalyst on Selective Methanation of CO in H2-Rich Gas
GAO Zhi-ming, DAI Qian-zi, MA Hong-wei
(School of Chemistry, Beijing Institute of Technology, Beijing 102488, China)
A series of 20%NiO(Clx)/CeO2samples with feed atomic ratio of Cl/Ce atx=0.0~0.30 were prepared by impregnation method and used to selective methanation of CO in H2-rich gas (CO-SMET) after reduction. It is found that chlorine doped suppressed CO2conversion greatly. And CO concentration was thus decreased to below 10-5over 20% Ni(Cl0.2)/CeO2catalyst with CO methanation reaction selectivity not less than 50% in a wide reaction temperature range of 220~240 ℃, meeting the requirement for fuel of proton exchange membrane fuel cell (PEMFC). In order to reveal effect of chlorine doping, measurements of X-ray diffraction (XRD), temperature programmed reduction (TPR) and adsorption isotherms of CO and CO2were conducted. Phase composition of the samples and crystallite size of CeO2were estimated by the Rietveld method. Thickness of crystallites of NiO or Ni was estimated by the Scherrer equation.
nickel; ceria; chlorine; CO selective methanation; hydrogen purification
2015-10-22
国家自然科学基金资助项目( 21171020)
高志明(1964—),男,教授,博士生导师,E-mail:zgao@bit.edu.cn.
O 643.322
A
1001-0645(2016)04-0429-07
10.15918/j.tbit1001-0645.2016.04.017
